Workflow Tutorial¶
This tutorial walks through a complete enzyme redesign session using CYP450 (PDB ID: 1SUO) as the case study. You will learn how to identify design hotspots, generate virtual saturation mutagenesis libraries, rationally evaluate mutants, cluster candidates, cross-screen with external tools, and explore co-evolution constraints.
Prerequisites¶
- REvoDesign installed and working in PyMOL
- Evolution data (PSSM + GREMLIN) pre-computed for your target sequence (see below)
Obtaining Evolution Data¶
REvoDesign requires PSSM and GREMLIN profiles computed from sequence databases. Use the computation service:
- Go to https://revodesign.yaoyy.moe/PSSM_GREMLIN/create_task
- Upload a FASTA-format sequence file (one sequence per file).
Sequences may contain unknown residues (
X) but not stop codons (*). - Monitor progress at the Dashboard.
- Hover over a task to reveal a cancel button (if queued/running) or a download button (if complete).
- When complete, download and unzip the archive for use in the Prepare step.
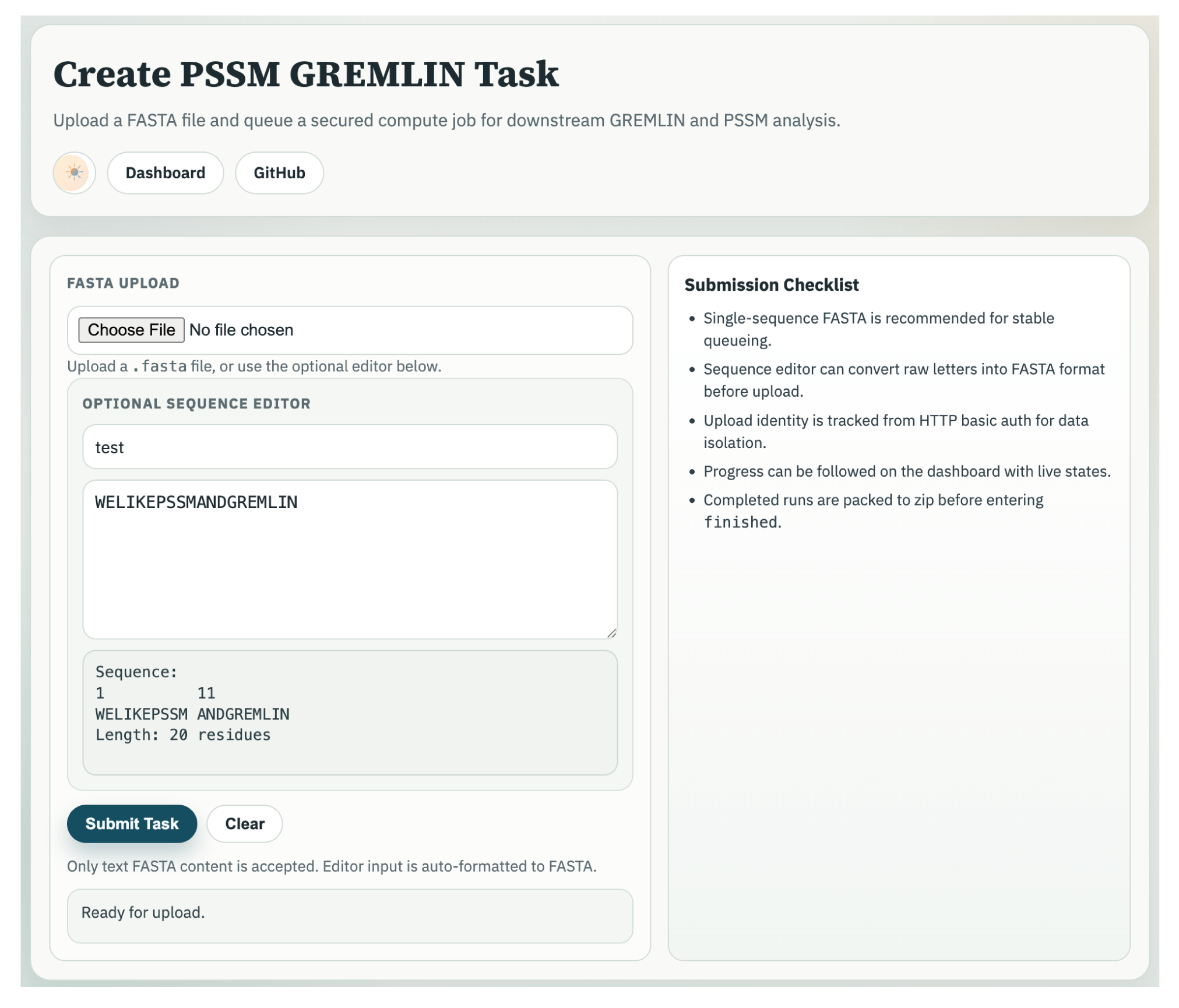
Example FASTA
>1SUO_A
XXXXXXXXXXXXXXXXXXXXXXXXXXXGKLPPGPSPLPVLGNLLQMDRKGLLRSFLRLREKYGDVFTVYLGSRPVVVLCGTDAIREALVDQAEAFSGRGKIAVVDPIFQGYGVIFANGERWRALRRFSLATMRDFGMGKRSVEERIQEEARCLVEELRKSKGALLDNTLLFHSITSNIICSIVFGKRFDYKDPVFLRLLDLFFQSFSLISSFSSQVFELFSGFLKYFPGTHRQIYRNLQEINTFIGQSVEKHRATLDPSNPRDFIDVYLLRMEKDKSDPSSEFHHQNLILTVLSLFFAGTETTSTTLRYGFLLMLKYPHVTERVQKEIEQVIGSHRPPALDDRAKMPYTDAVIHEIQRLGDLIPFGVPHTVTKDTQFRGYVIPKNTEVFPVLSSALHDPRYFETPNTFNPGHFLDANGALKRNEGFMPFSLGKRICLGEGIARTELFLFFTTILQNFSIASPVPPEDIDLTPRESGVGNVPPSYQIRFLARH
Step 1: Prepare the Structure¶
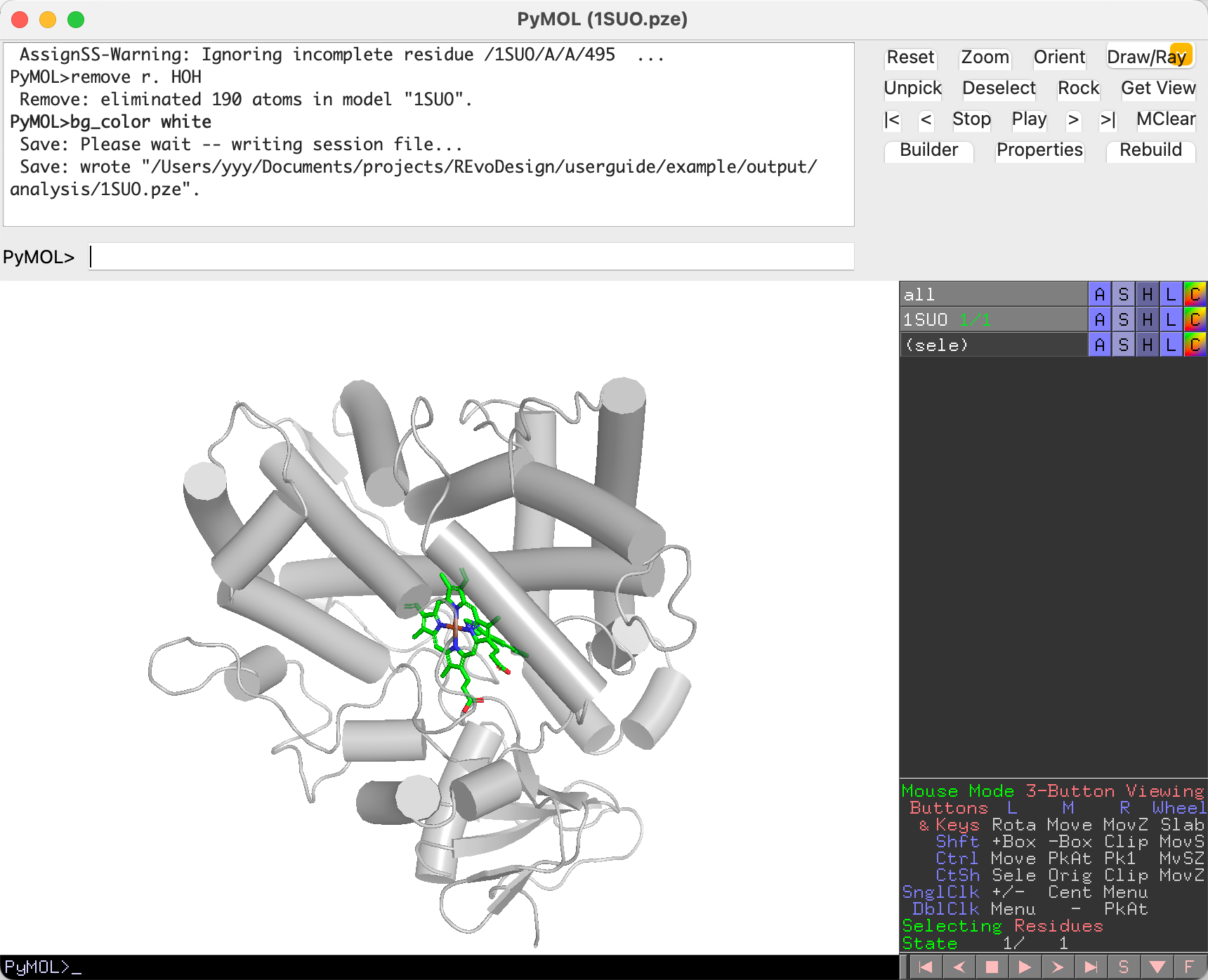
Load and Set Up in PyMOL¶
In PyMOL, fetch the target structure and prepare it for analysis:
fetch 1SUO
1SUO is a CYP450 enzyme with four components:
| Segment ID | Molecule | Description |
|---|---|---|
| A | Protein | Enzyme |
| B | HEM | Cofactor |
| C | CPZ | Substrate |
| D | HOH | Crystallization water |
Apply basic styling and clean up:
# Cartoon styling
set cartoon_cylindrical_helices, 1
set cartoon_color, gray70
set cartoon_transparency, 0.3
# Fix secondary structure assignment
dss
# Remove crystallization water
remove resn HOH
# White background
bg_color white
Save this session — it will serve as the starting point for all subsequent analysis.
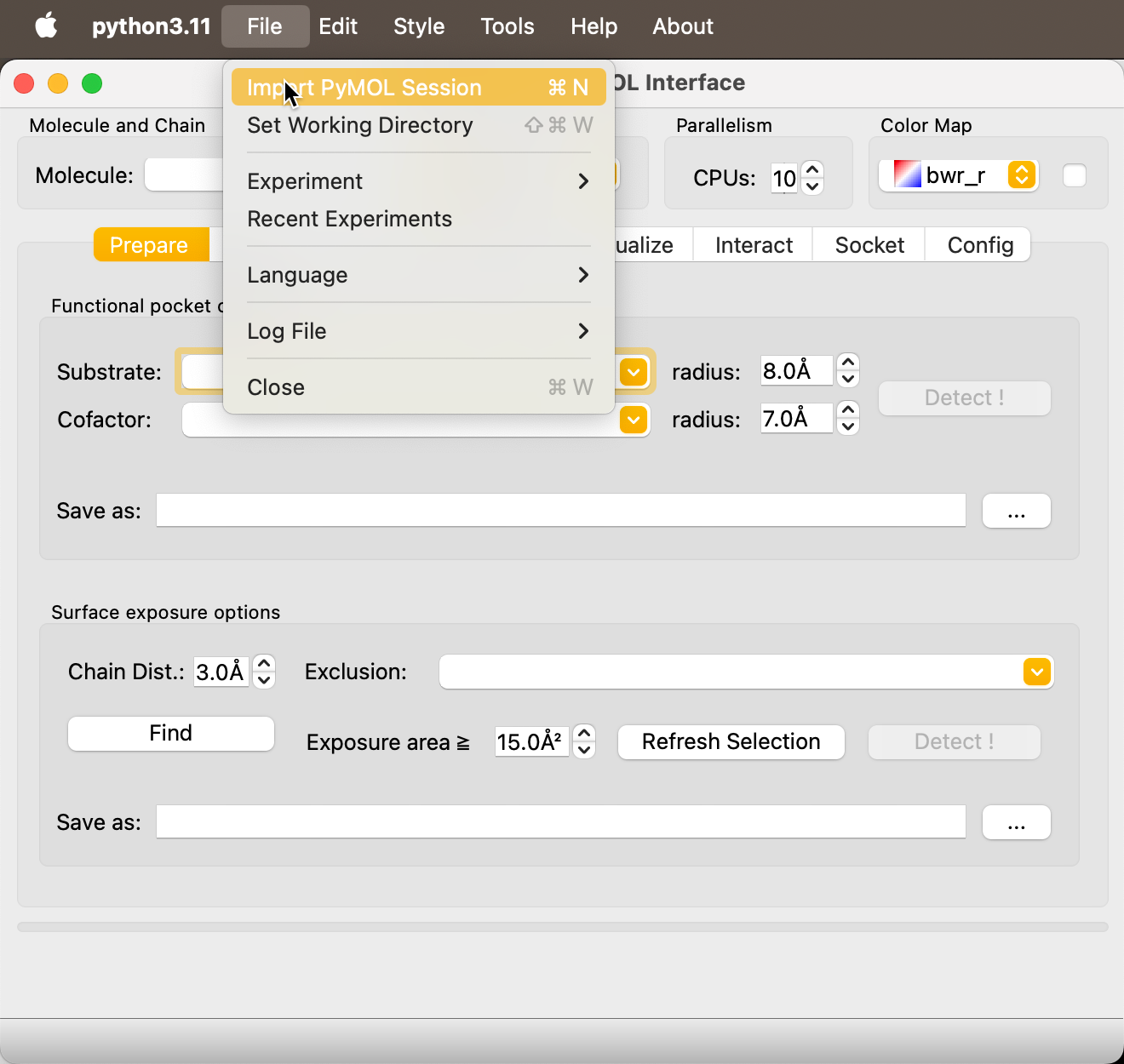
Import Session into REvoDesign¶
In REvoDesign, go to File → Import PyMOL Session (or press
Ctrl+N / Cmd+N). This registers the PyMOL session so REvoDesign can
identify molecules and selections.
Detect Binding Pocket Hotspots¶
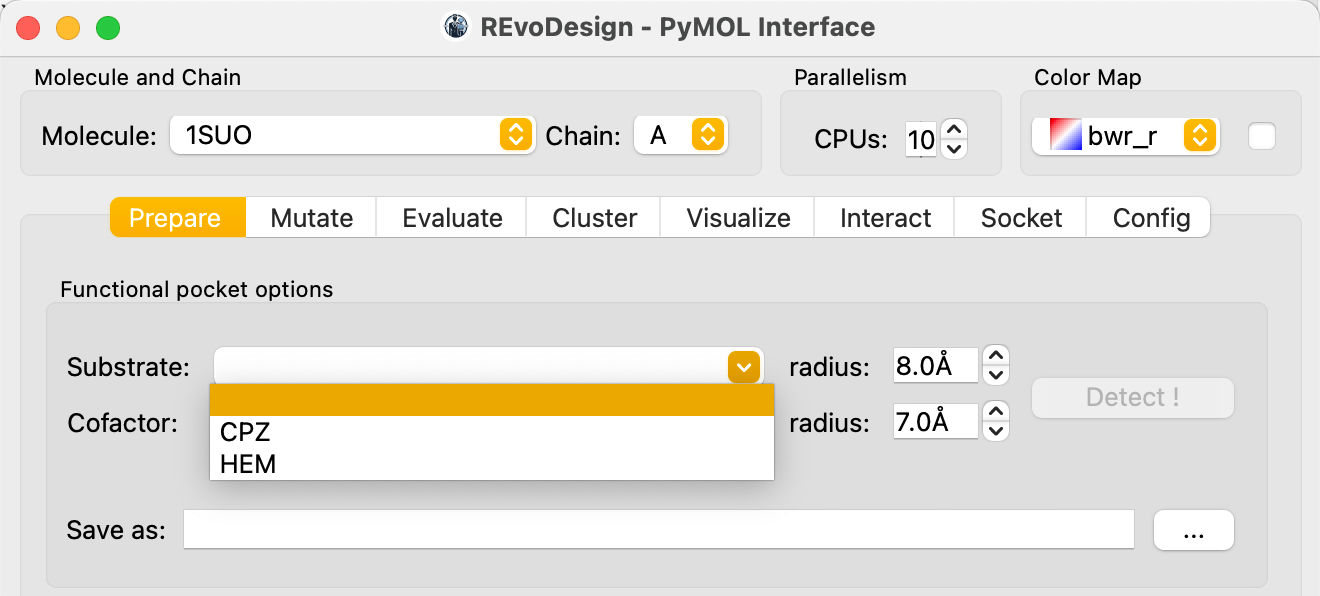
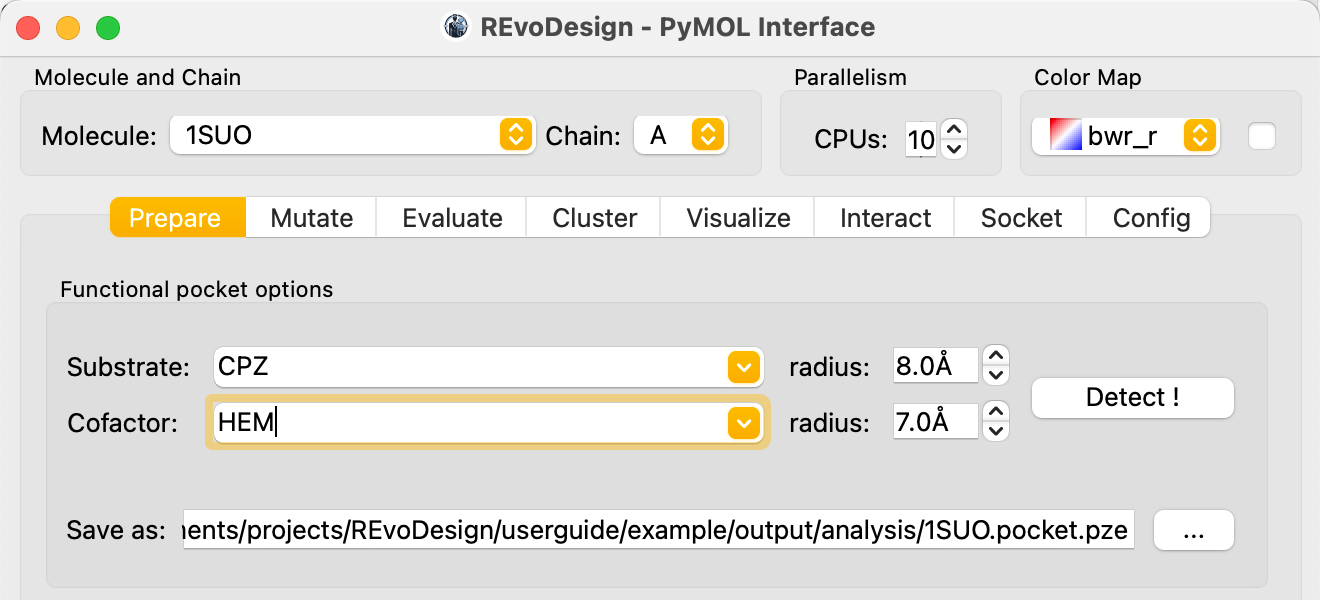
In the Prepare tab, under the Pocket section:
- Identify the substrate and cofactor molecules by their residue names
(e.g.,
CPZfor the substrate,HEMfor the cofactor). - Set the contact distance cutoff for each (default: 8 Å for substrate, 7 Å for cofactor).
- Specify a save path to enable the detection button.
- Click Detect.
You can also use PyMOL selection syntax for complex cases (e.g.,
r. UNK or r. LIG to treat two ligands as one).
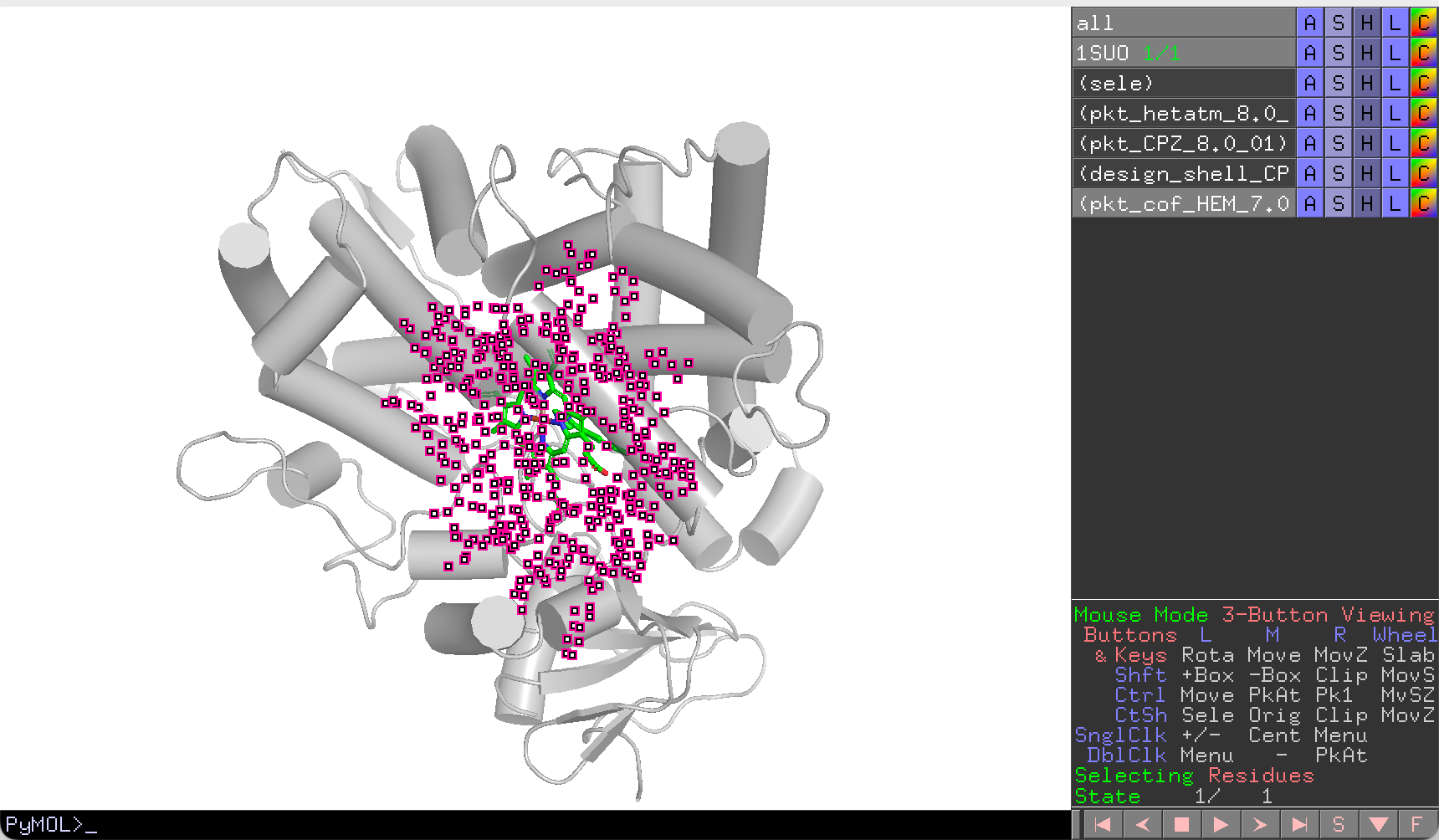
How pocket detection works
Residues within the cutoff distance of the substrate/cofactor are collected into selection groups. Overlapping regions (shared by cofactor and substrate) are assigned to the cofactor group and removed from the substrate group to avoid double-counting.
Results are saved to pockets/ in the working directory as
<molecule>_<pocket_selection>_residues.txt. The following selections
are created:
| Selection | Content |
|---|---|
design_shell_CPZ_8.0_01 |
Substrate-binding residues (cofactor overlap removed) |
pkt_cof_HEM_7.0_01 |
Cofactor-binding residues |
pkt_CPZ_8.0_01 |
Substrate-binding residues (full) |
pkt_hetatm_8.0_01 |
All heteroatom-contacting residues (union) |
Detect Surface-Exposed Hotspots¶
In the Prepare tab, under the Surface Exposure section:
- Set the solvent-accessible surface area (SASA) threshold (default: 15 Ų). Residues with SASA ≥ threshold are considered surface-exposed.
- Optionally exclude pocket residues: click Refresh Selection to load
available PyMOL selections, then choose
pkt_hetatm_8.0_01from the Exclusion dropdown. - Specify a save path and click Find.
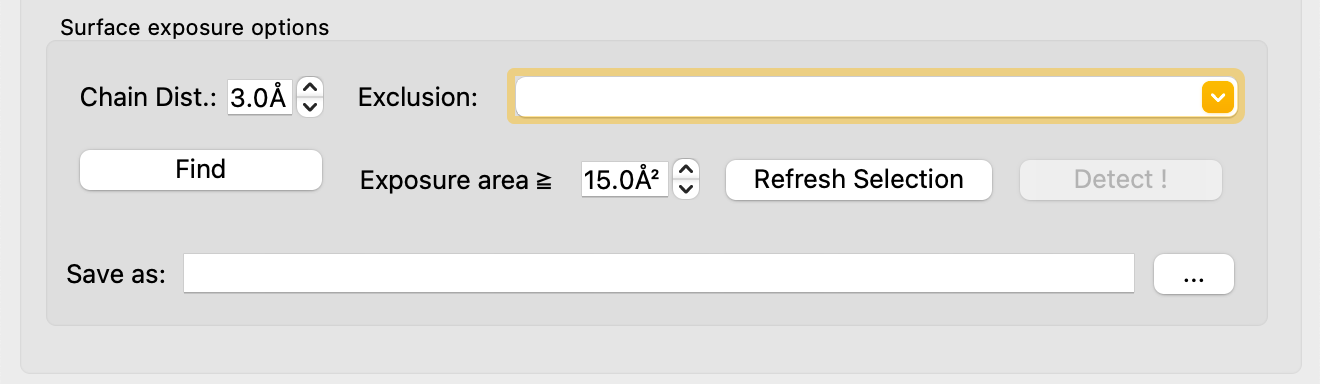
Results are visualized as spheres: blue for exposed, red for buried,
and no sphere for excluded residues. Results are also saved to
surface_residue_records/ in the working directory.
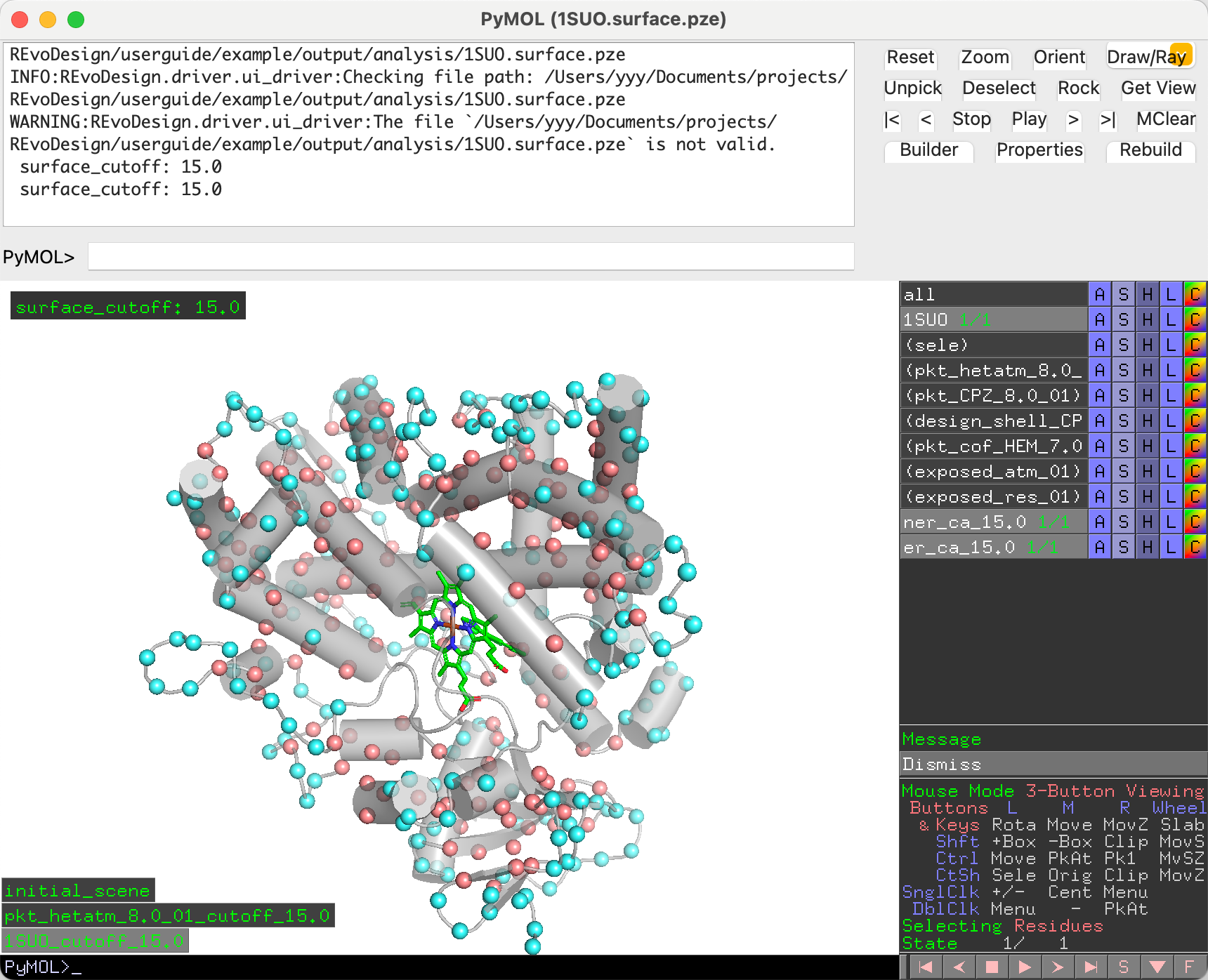
Warning
The surface-exposure visualization session is for inspection only. Do not use it as the basis for further design steps — use the pre-detection session instead.
Protein-Protein Interface (Optional)¶
For multimeric proteins, use the PPI section to detect chain-chain contacts:
- Set Chain Dist to the minimum contact distance between chains.
- Click Find to identify interfacial residues.
- Click Refresh Selection to load the result for exclusion.
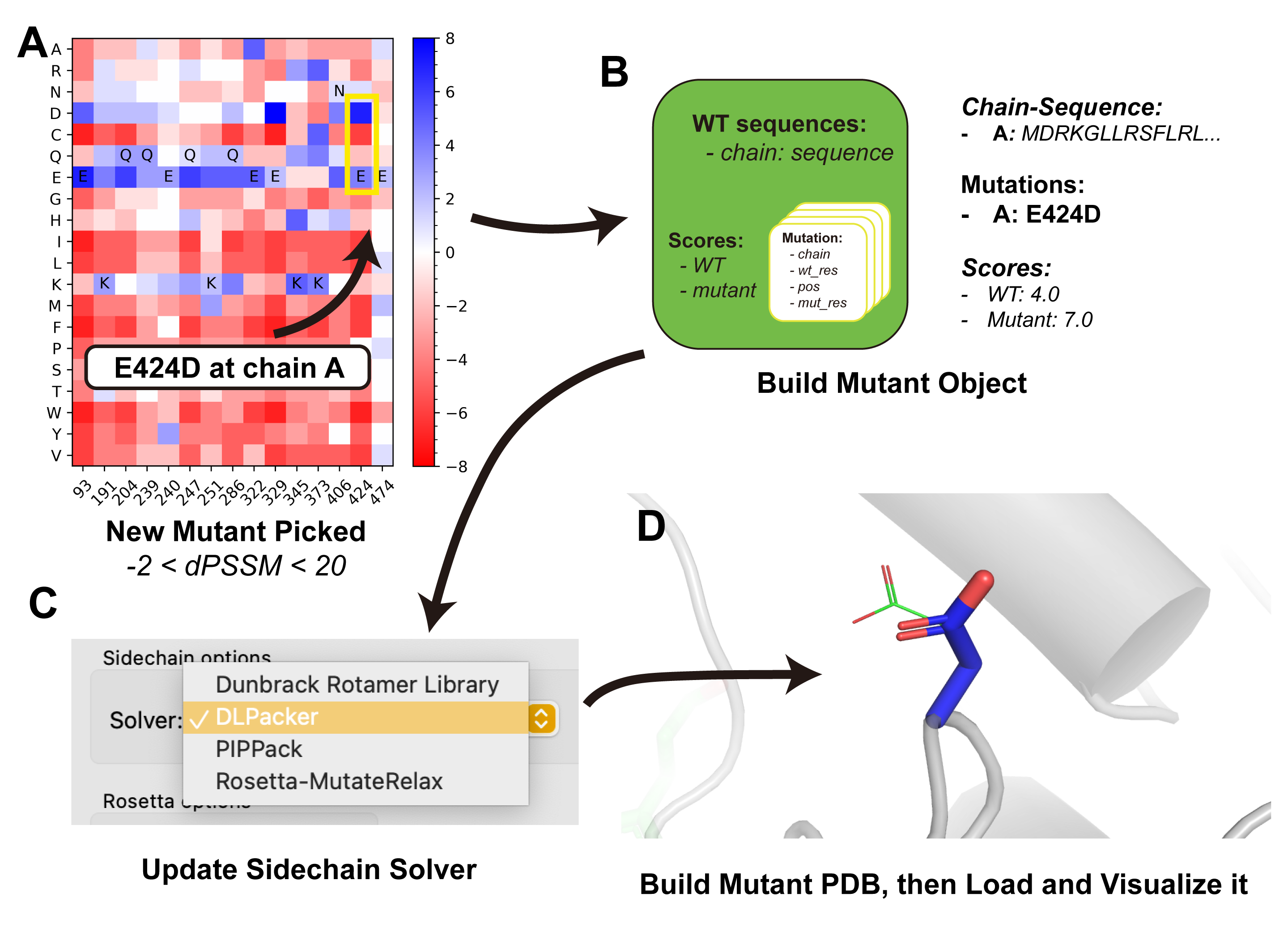
Step 2: Mutate — Virtual Saturation Mutagenesis¶
The Mutate tab generates a pool of virtual point mutations under constraints derived from evolutionary conservation (PSSM).
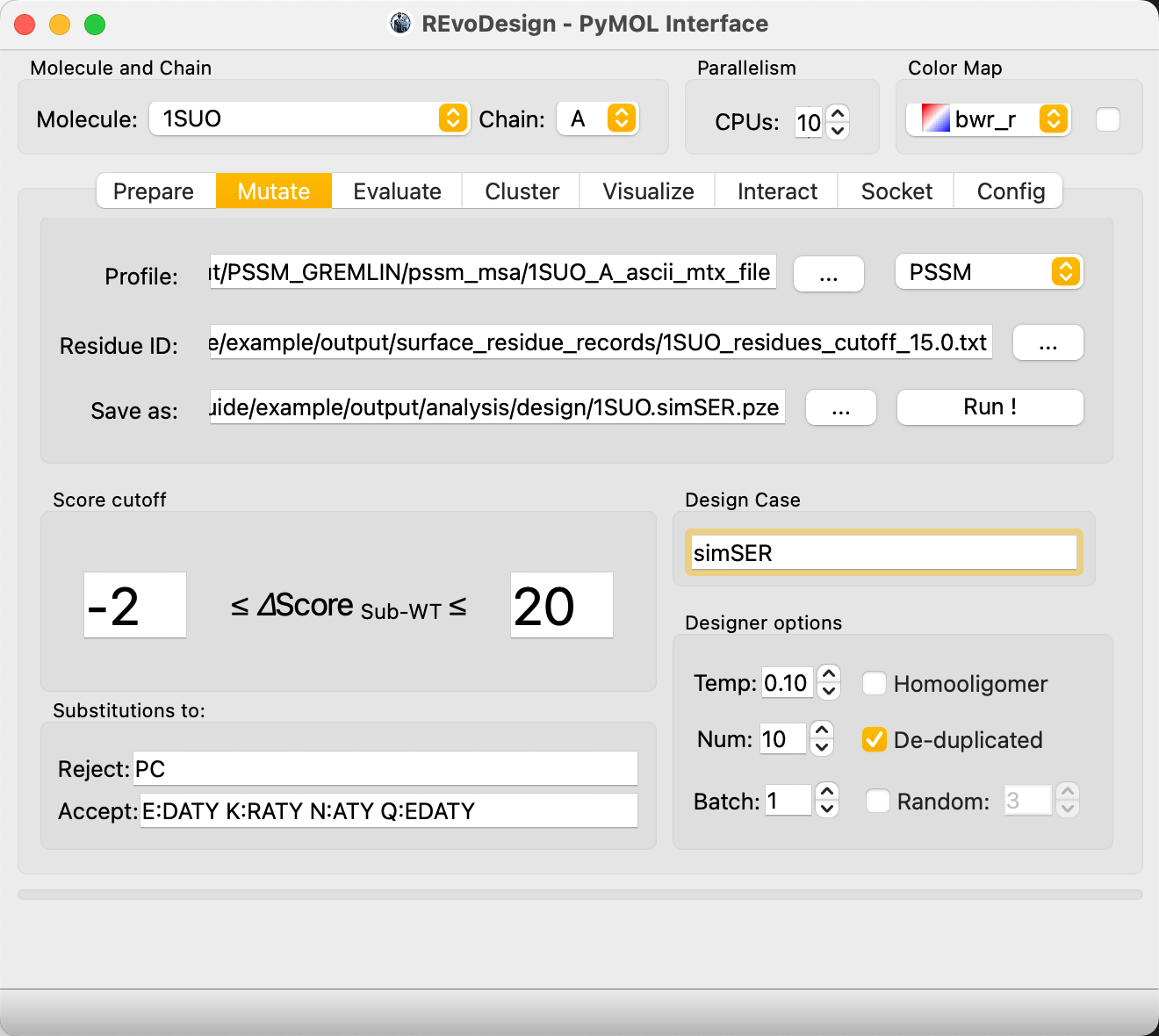
Strategy 1: Surface Entropy Reduction¶
Surface entropy reduction replaces exposed residues with shorter, less solvent-interacting amino acids within conservation constraints.
- Load the unzipped evolution data into Profile and set type to PSSM.
- Set Residue ID to the surface exposure result selection.
- Choose a session save path.
- Set Score cutoff bounds. Example: PSSM score difference ≥ -2 and ≤ 20 relative to wild-type (-2 tolerates slightly less conserved substitutions; 20 is effectively unbounded, meaning absolute conservation).
- In Substitution:
- Reject:
PC(reject proline and cysteine) - Accept: e.g.,
E:DATY(replace E with D/A/T/Y candidates)
- Reject:
- Enter a Design Case name for output file naming.
- Click Run!
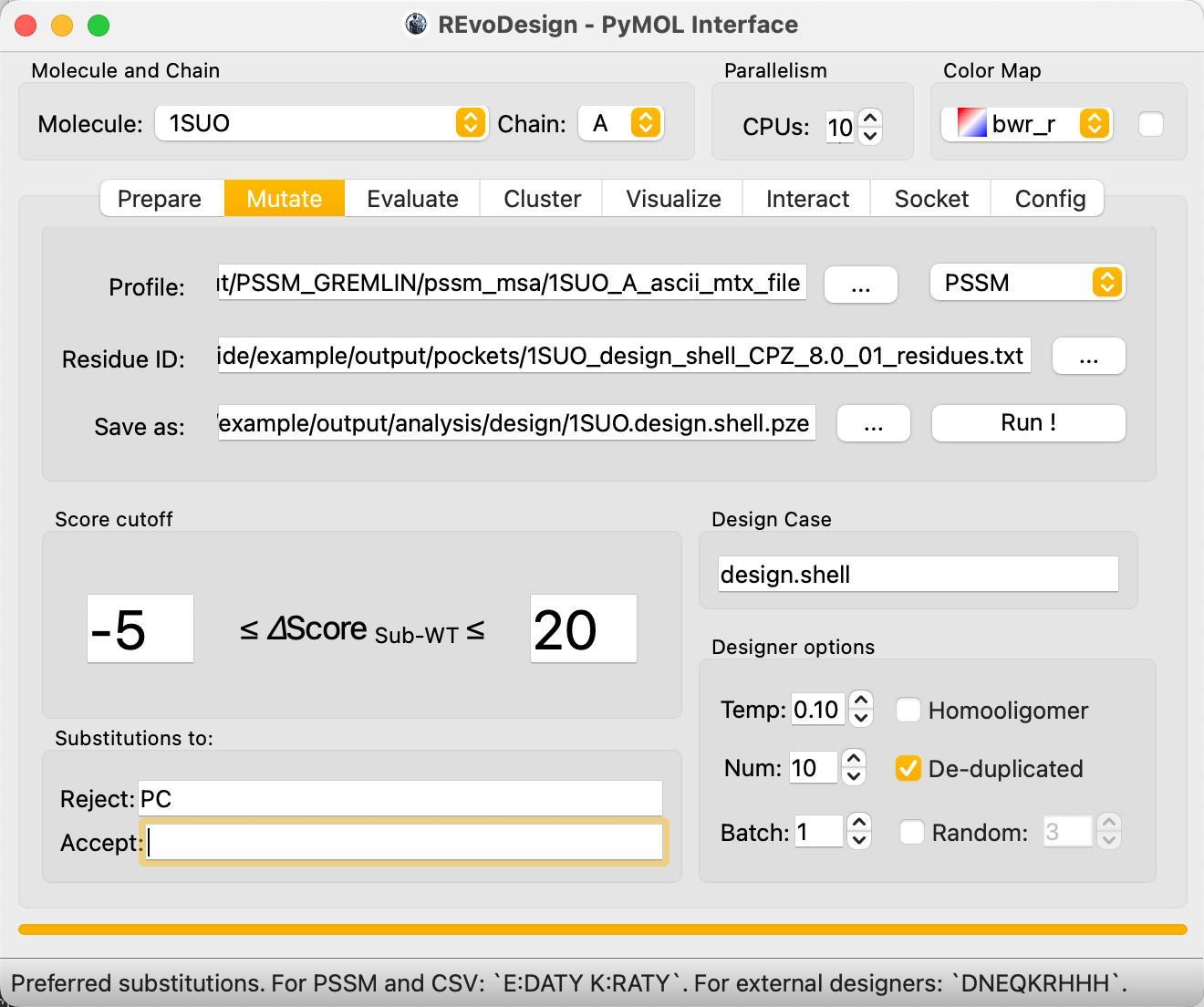
Strategy 2: Catalytic Pocket Design¶
Catalytic pocket design uses a more permissive substitution strategy to increase diversity near the active site.
- Set Score cutoff to a wider range (e.g., ≥ -5, ≤ 20).
- Clear the Accept substitution preferences to allow all valid substitutions.
- Keep Reject as
PCto avoid disruptive proline/cysteine mutations.
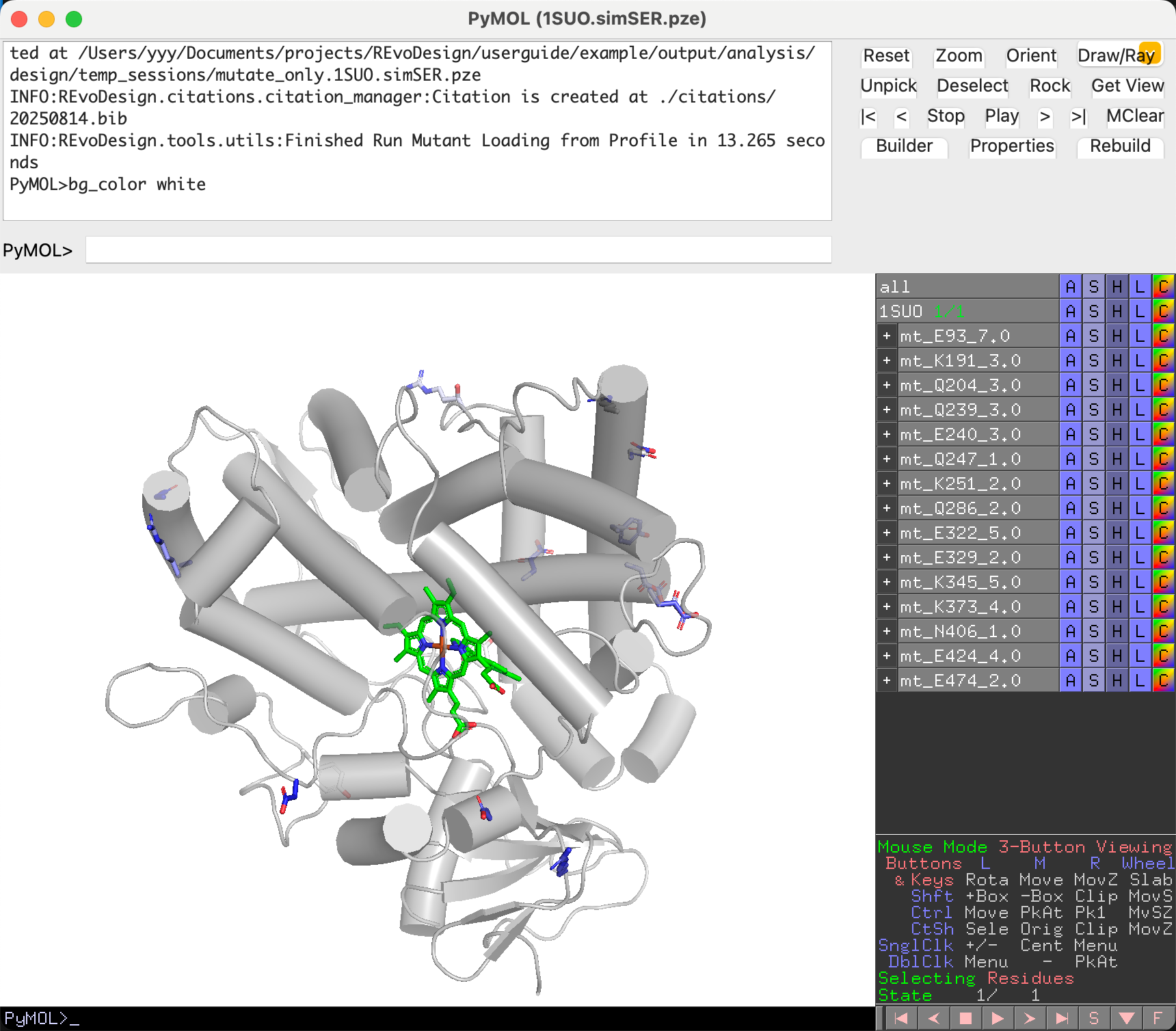
Understanding the Output¶
Design results appear in PyMOL grouped by residue position:
- Group name:
mt_<WT><position>_<PSSM_score> - Mutant name:
<chain><WT><position><mutant>_<mutant_score> - Only the mutated sidechain is shown
- Carbon atoms are colored by PSSM score (see color preset)
- Full PDB structures are saved under
mutant_pdbs/in the working directory
Step 3: Evaluate — Rational Screening¶
The Evaluate tab provides tools for visual, side-by-side comparison of wild-type and mutant sidechains to make informed decisions.
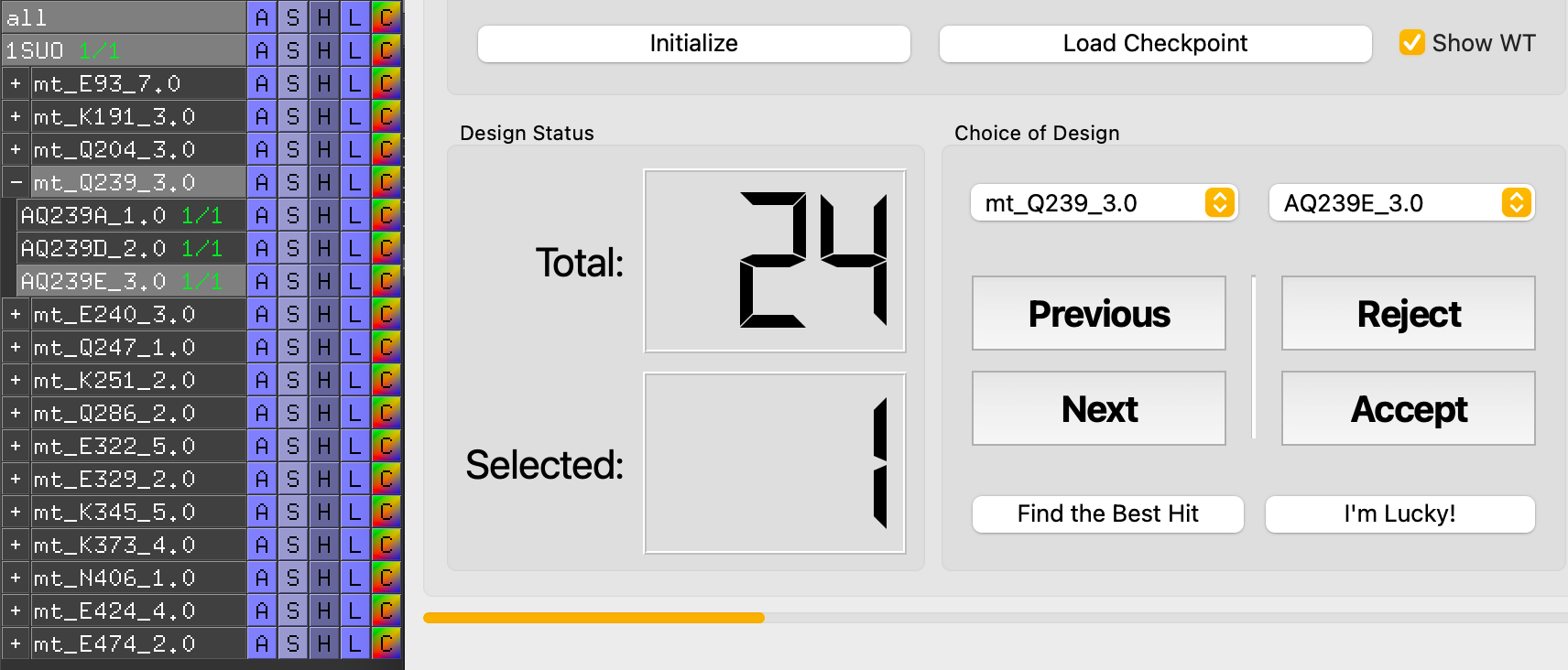
Initialize the MutantTree¶
REvoDesign organizes mutants into a MutantTree — branches are residue positions, leaves are individual point mutants at that position.
- Go to the Evaluate tab.
- Set a save path for mutant records and checkpoint files.
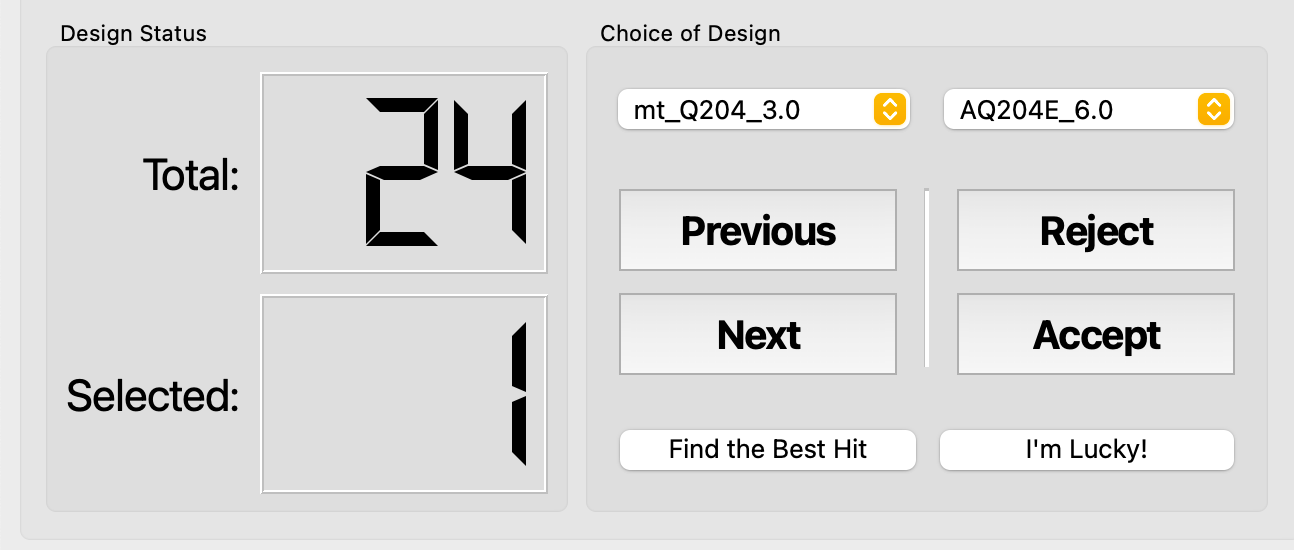
- Click Initialize to scan the PyMOL session for mutant trees. If successful, Total shows a non-zero count and decision buttons become enabled.
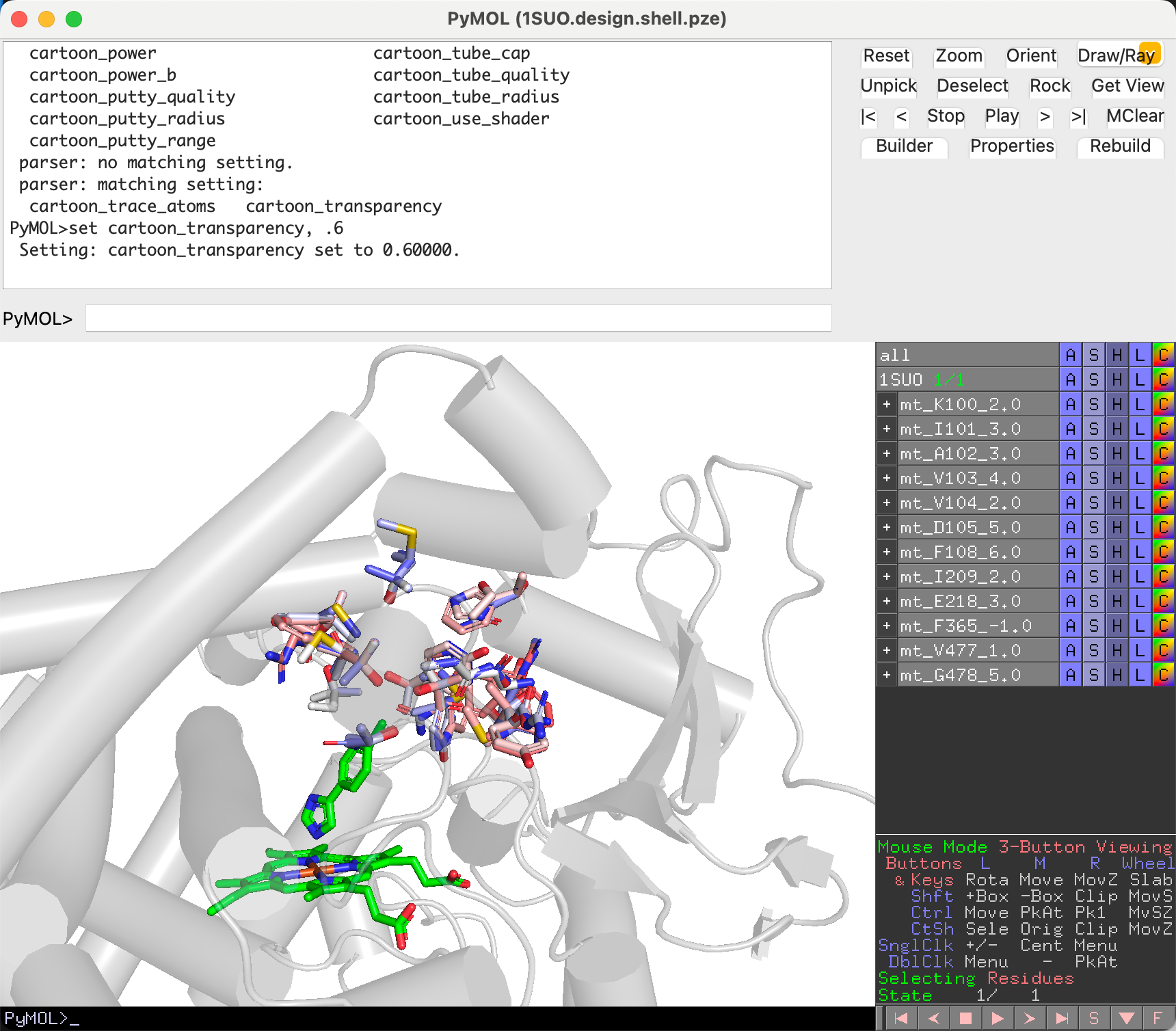
Navigate and Decide¶
In evaluation mode, REvoDesign hides all other mutants, collapses unrelated branches, and shows only the current branch and individual. The wild-type sidechain is displayed as a wireframe for comparison, while the mutant sidechain is shown in ball-and-stick with a mesh surface.
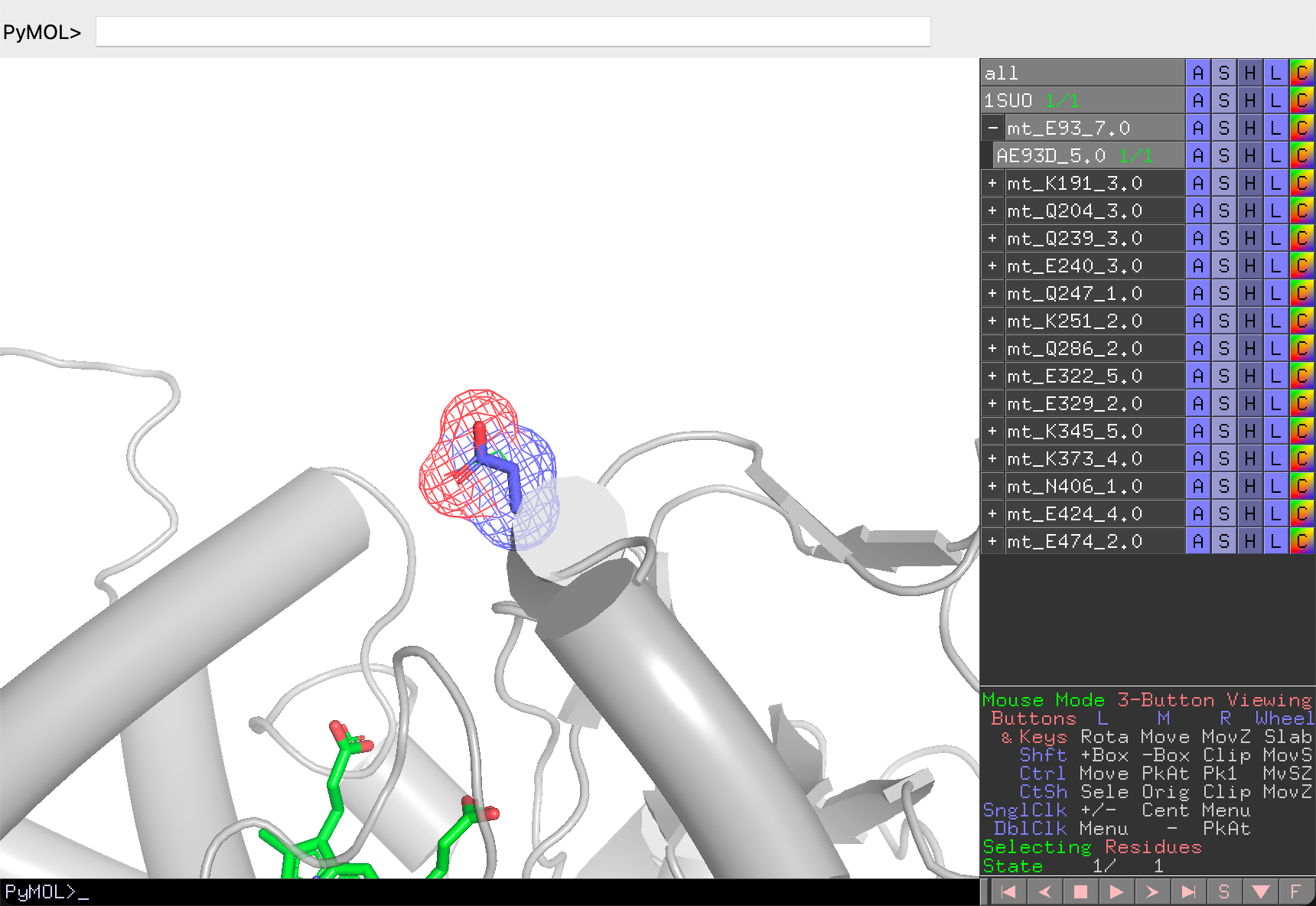
Decision actions:
| Button | Action | Description |
|---|---|---|
| Previous | Go to previous mutant | Tooltip: Shift+Opt+[ |
| Next | Go to next mutant | Tooltip: Shift+Opt+] |
| Accept | Accept current mutant | Tooltip: Shift+Opt+- |
| Reject | Reject current mutant | Tooltip: Shift+Opt++ |
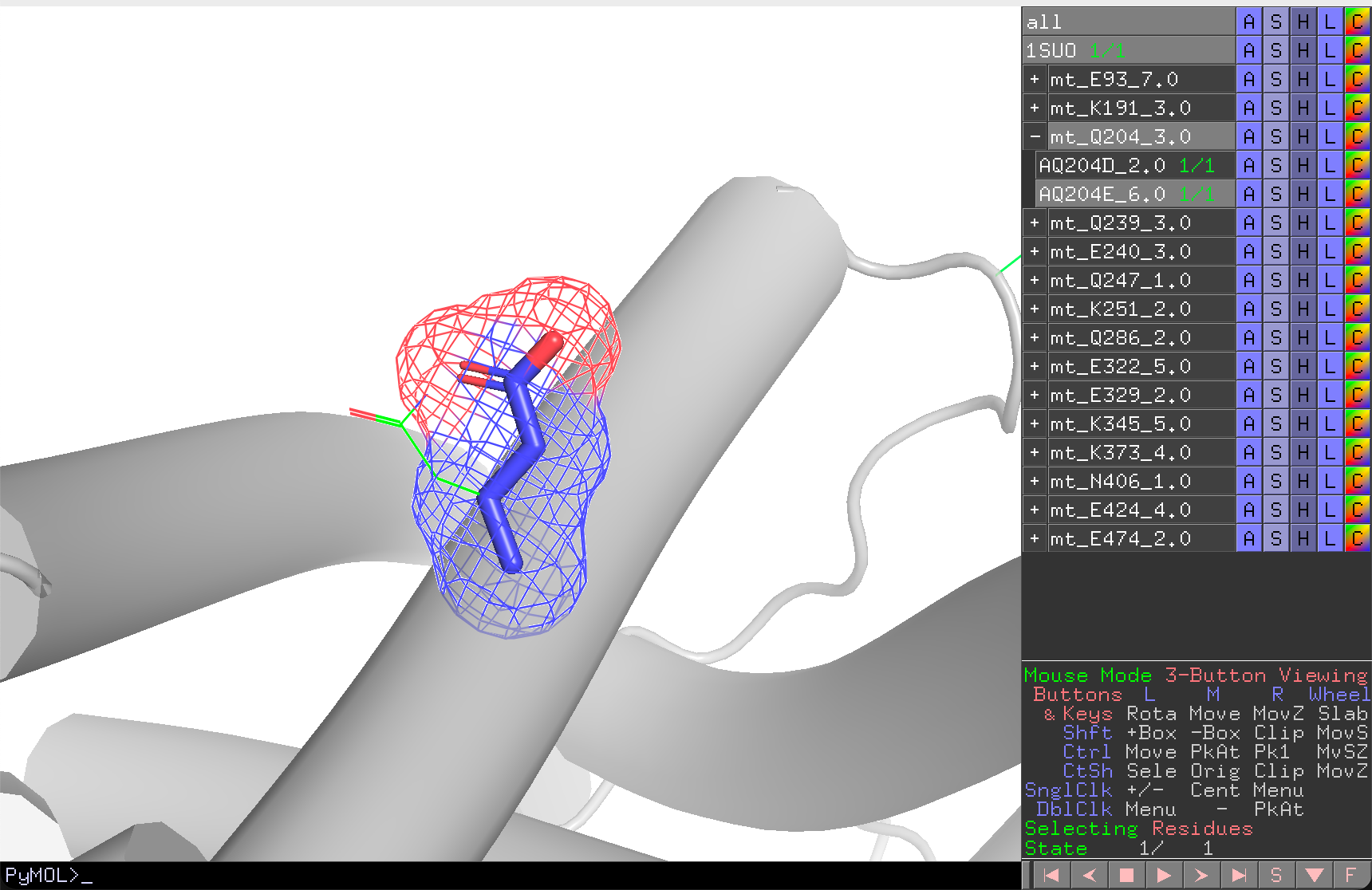
Fast Navigation¶
Use the dropdown menus to jump directly to a specific branch or mutant.
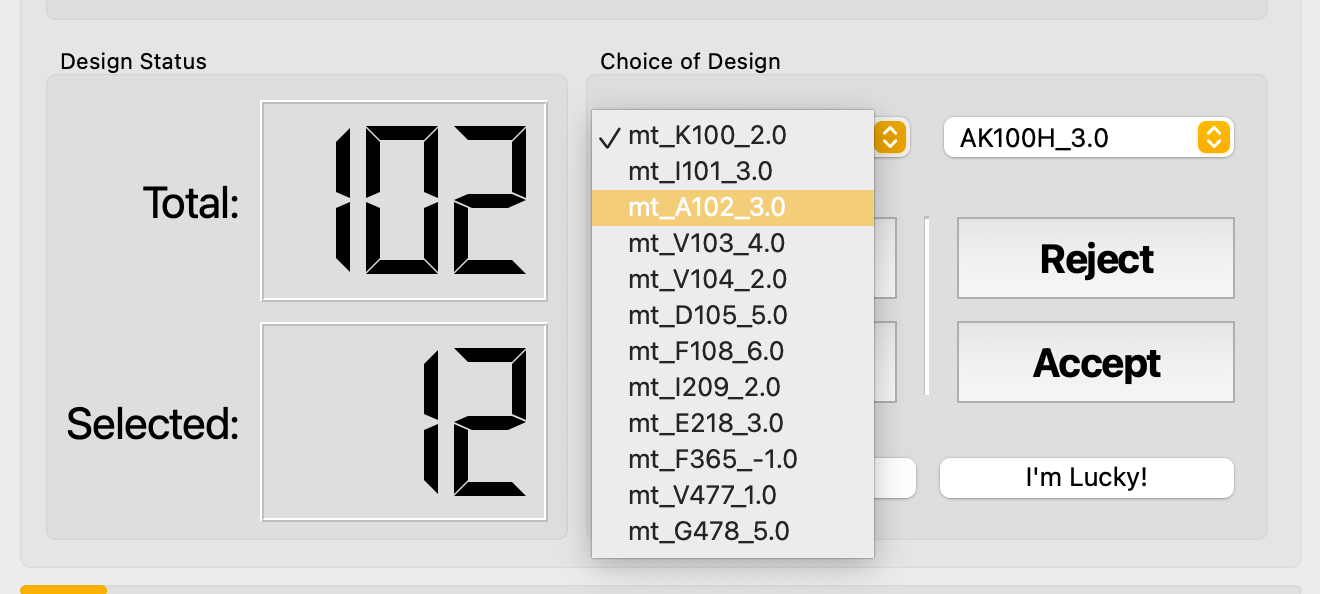
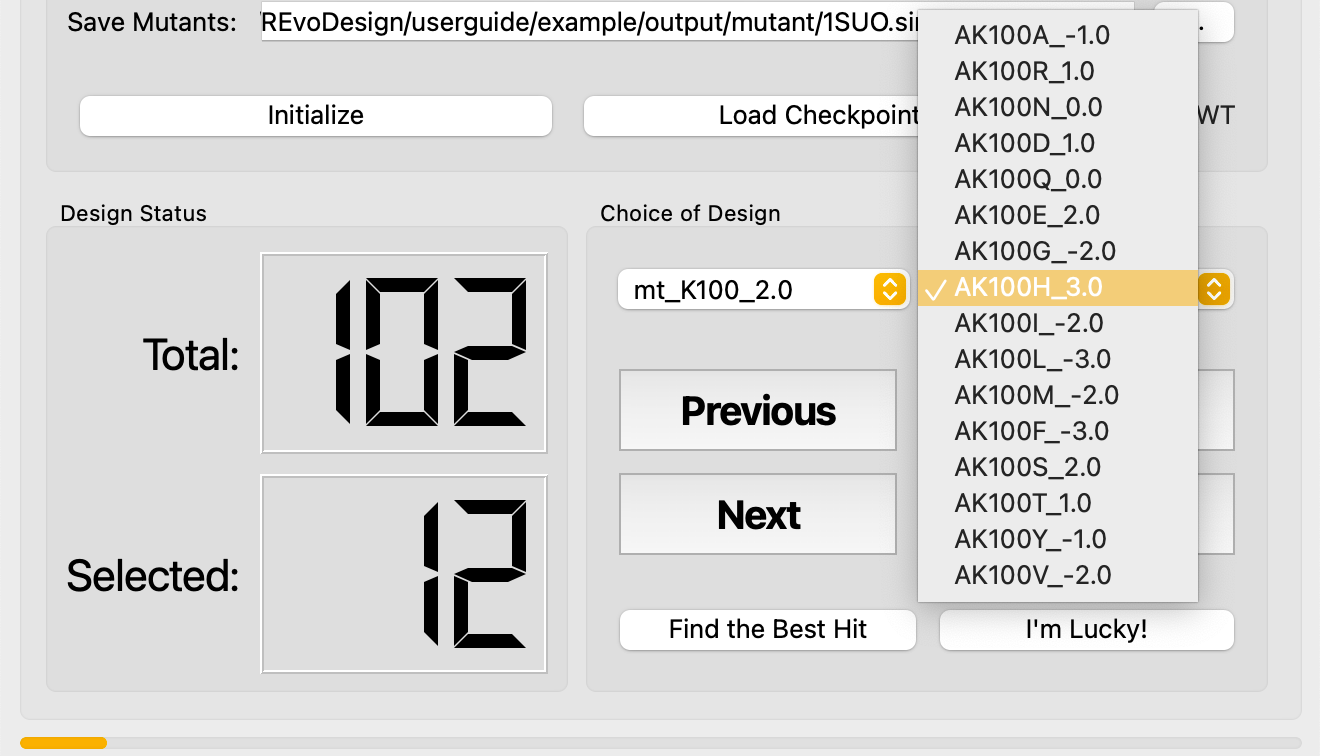
- Find the Best Hit — automatically jumps to the highest-scoring mutant in the current branch.
- I'm Lucky! — scans every branch and collects the highest-scoring mutant from each. This is a rapid way to identify promising leads across all positions.
Decision Persistence¶
Decision results are saved in real time to a text file, with corresponding checkpoint files for reloading.
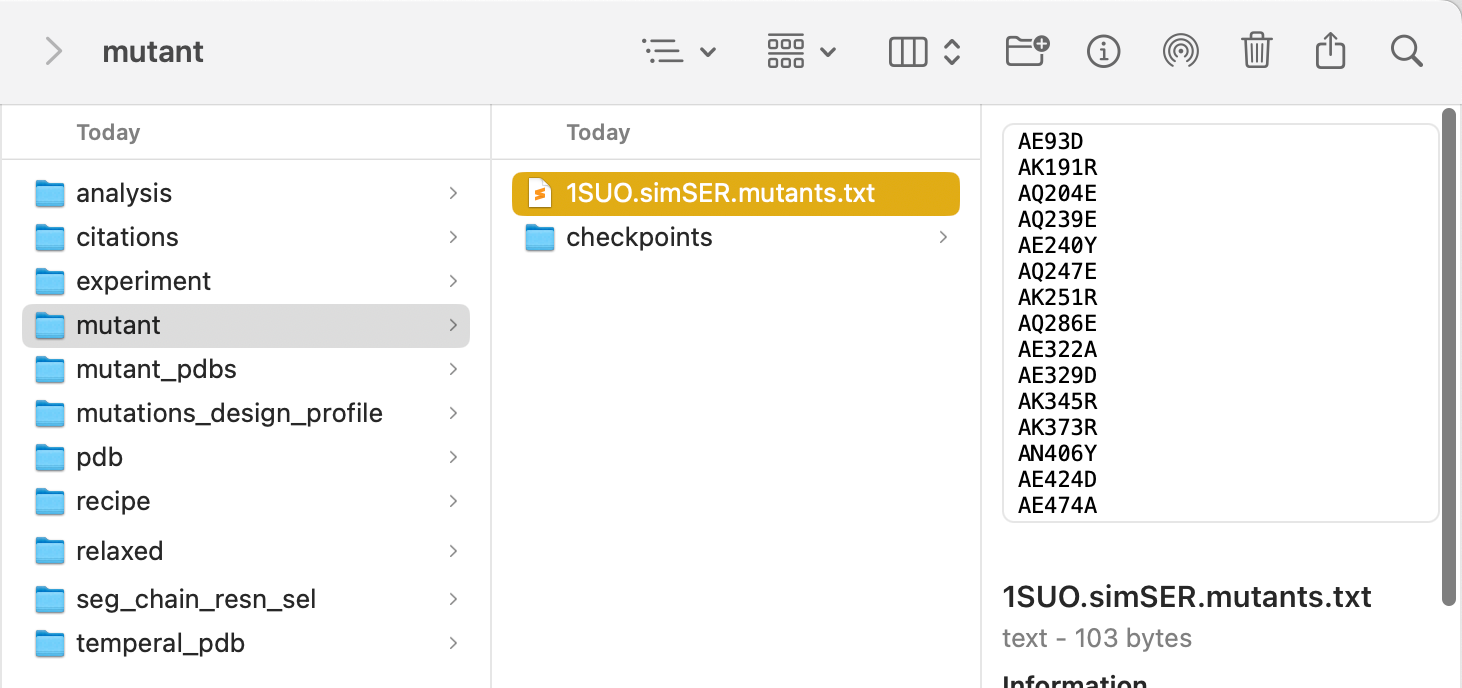
To reload a previous checkpoint:
- Re-initialize the MutantTree (clears previous decisions).
- Load the checkpoint file.
- Previous decisions are restored.
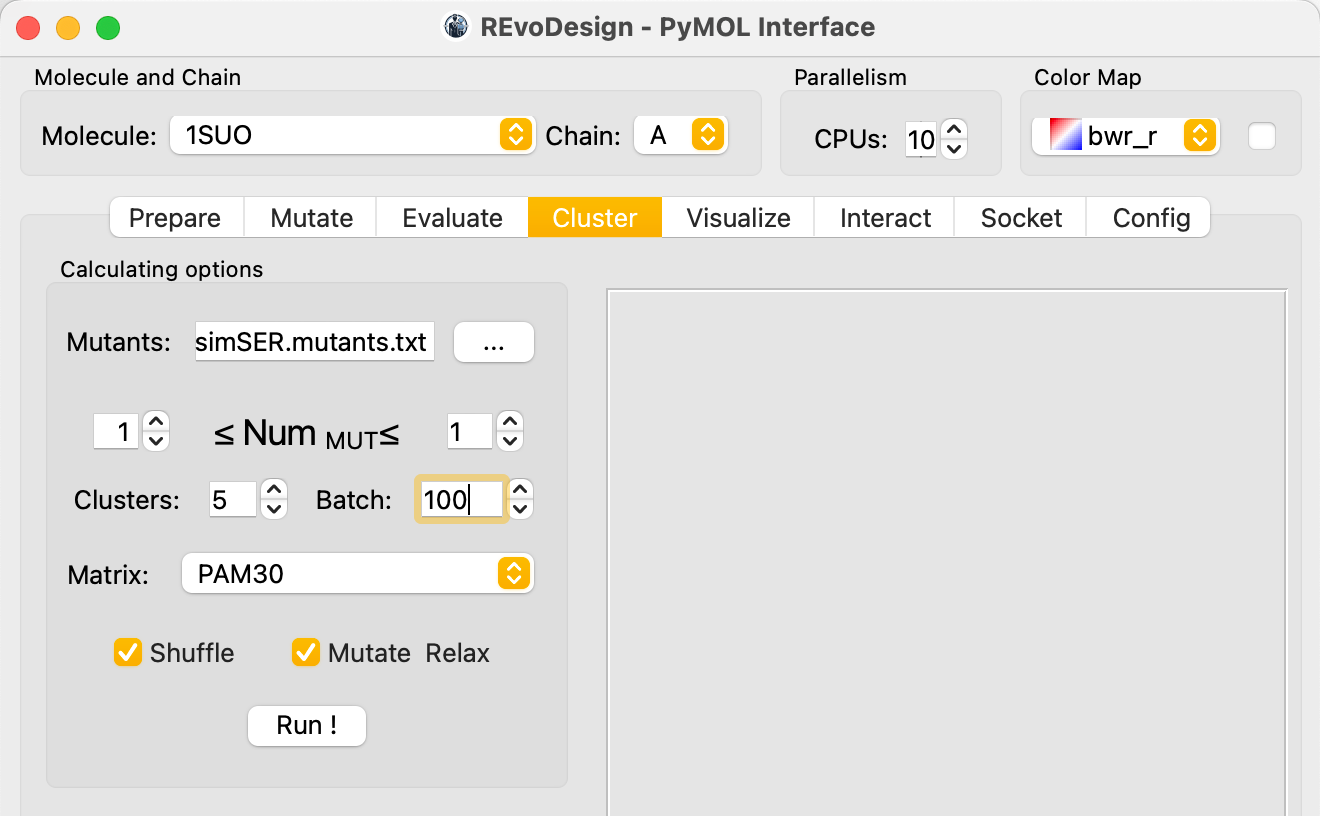
Step 4: Cluster — Reduce Library Size¶
REvoDesign uses sequence-based clustering to group similar mutants and select representatives from each cluster, reducing the library to a size manageable for wet-lab validation.
- Load the accepted mutant list from the Evaluate step.
- Set the number of mutations per mutant (default: 1).
- Set the number of clusters (must be less than total mutants).
- Choose a scoring matrix (default: PAM30).
- Optionally enable Mutate Relax to score representatives with Rosetta energy evaluation.
- Click Run.
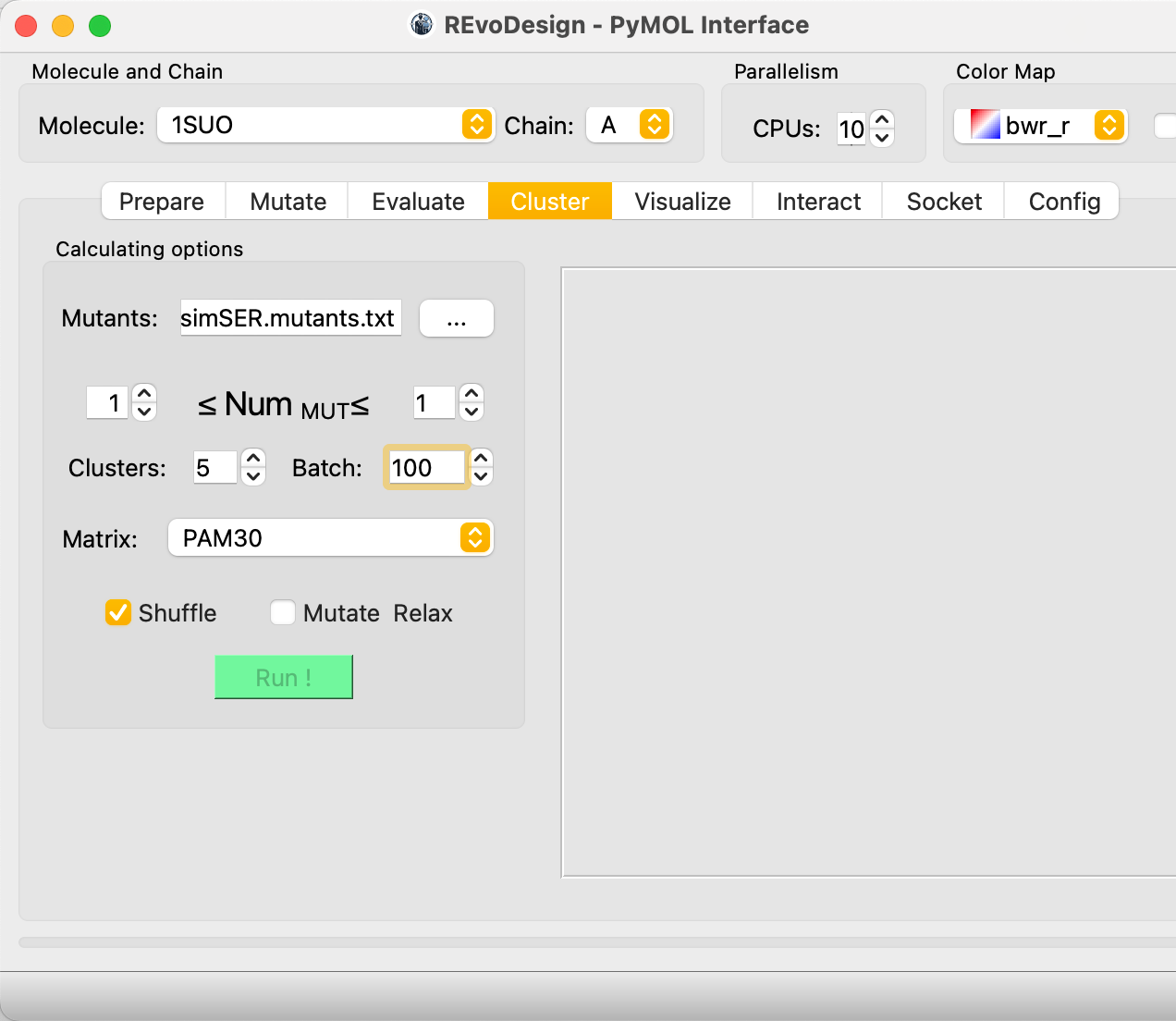
The results panel shows a pairwise sequence similarity matrix (darker = more similar).
Cluster count
Too few clusters can force unrelated sequences into the same group, masking diversity. Choose a cluster count that balances library size with sequence diversity preservation.
With Rosetta Energy Evaluation¶
When Mutate Relax is enabled, REvoDesign builds each mutant structure with Rosetta and evaluates its energy. The lowest-energy mutant in each cluster is selected as the representative.

Full scoring results are saved as both Excel and CSV files for downstream analysis.
Mutate Relax assumptions
Mutate Relax operates under three assumptions:
- The starting structure is already energy-minimized.
- Point mutations do not affect backbone coordinates.
- Point mutations do not affect distant sidechain packing.
Under these assumptions, only the mutated site is repacked locally. A well-optimized starting structure is critical for reliable scores.
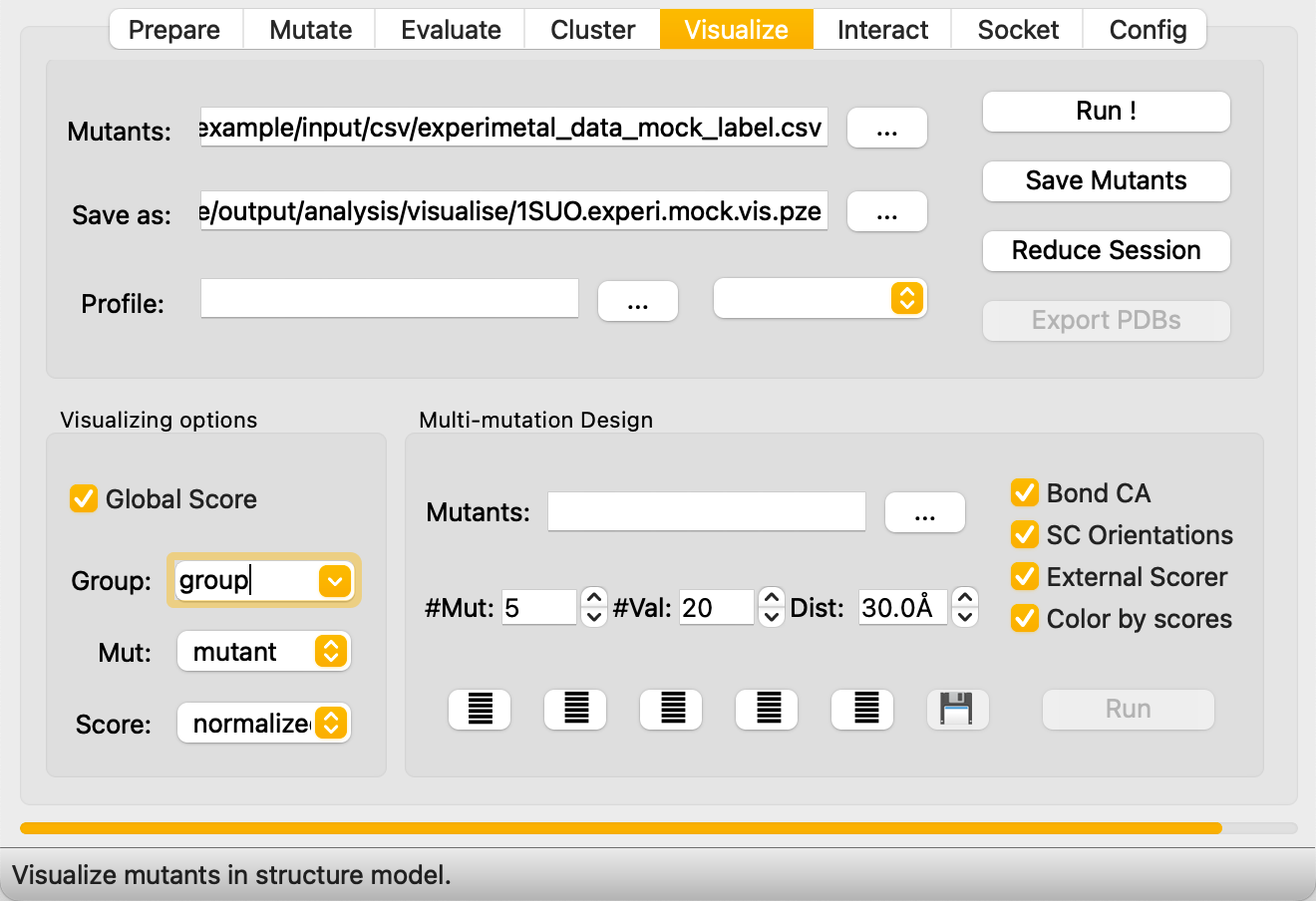
Step 5: Visualize — Cross-Screening and Data Display¶
The Visualize tab has two main functions:
Cross-Screening with External Scoring Tools¶
Combine REvoDesign's mutation list with scores from external tools (ddG predictors, stability predictors, etc.) for multi-criteria filtering.
This example uses Pythia-ddG, a structure-based ΔΔG predictor available on BioLib at https://biolib.com/YaoYinYing/pythia-wubianlab/.
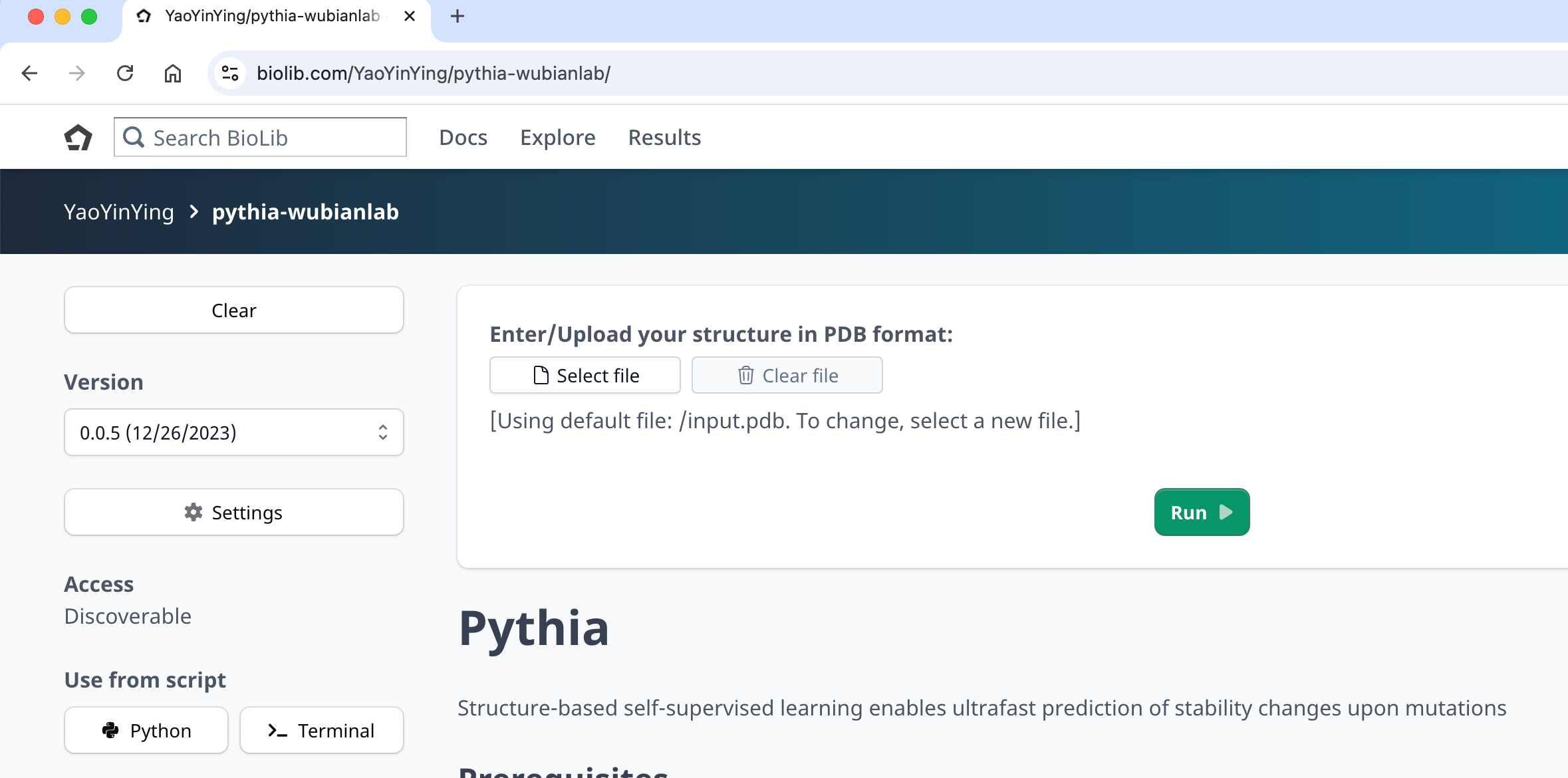
- Upload the PDB structure to Pythia-ddG and run (takes ~1 minute).
- Download the CSV results.
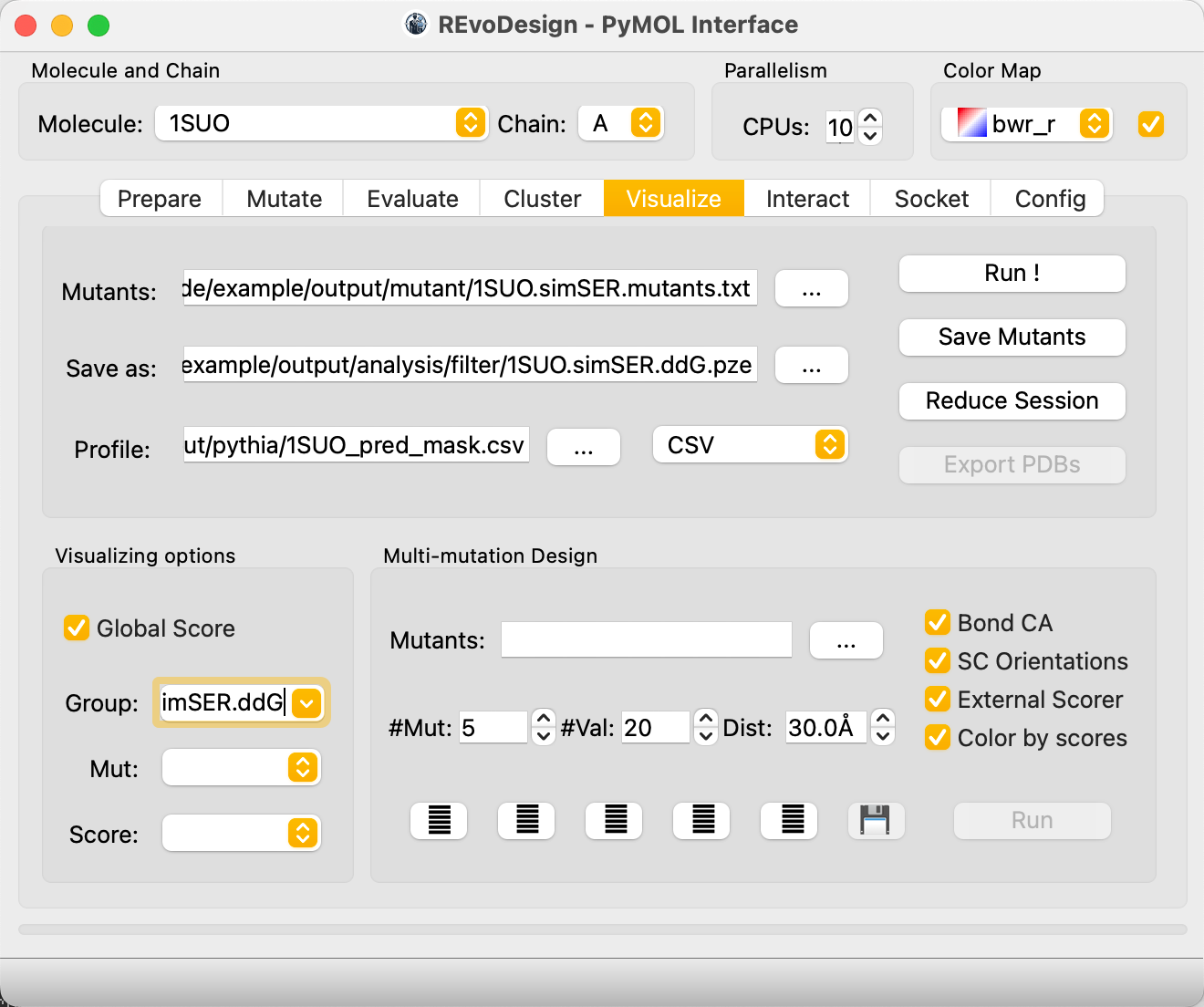
- In REvoDesign's Visualize tab:
- Load the mutant list.
- Set the save path.
- Select the Pythia-ddG CSV as the profile.
- Verify Profile type is set to
CSV. - Check Invert color preset (lower ddG = more stable = better).
- Check Global scoring to use full-table extremes for coloring.
- Enter a Group name for MutantTree organization.
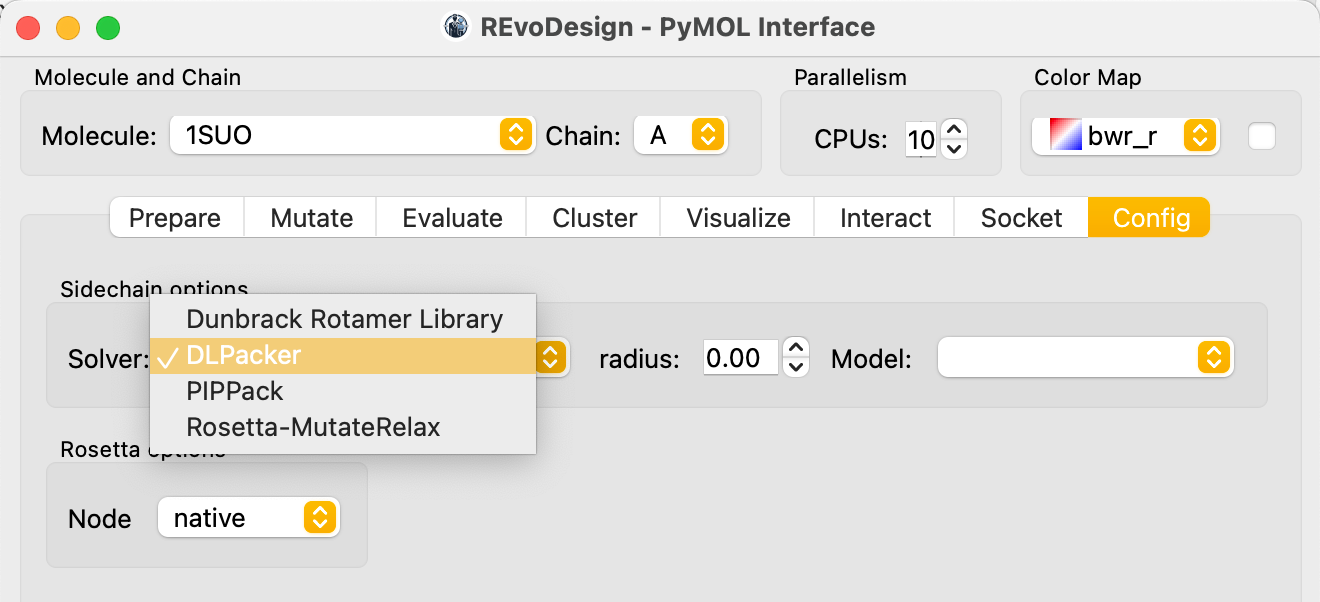
Sidechain solver for cross-screening
When building mutant structures for cross-screening, use a high-accuracy sidechain solver like DLPacker for reliable structural details during visual inspection.
Pruning the MutantTree¶
Unwanted mutants can be removed during cross-screening review:
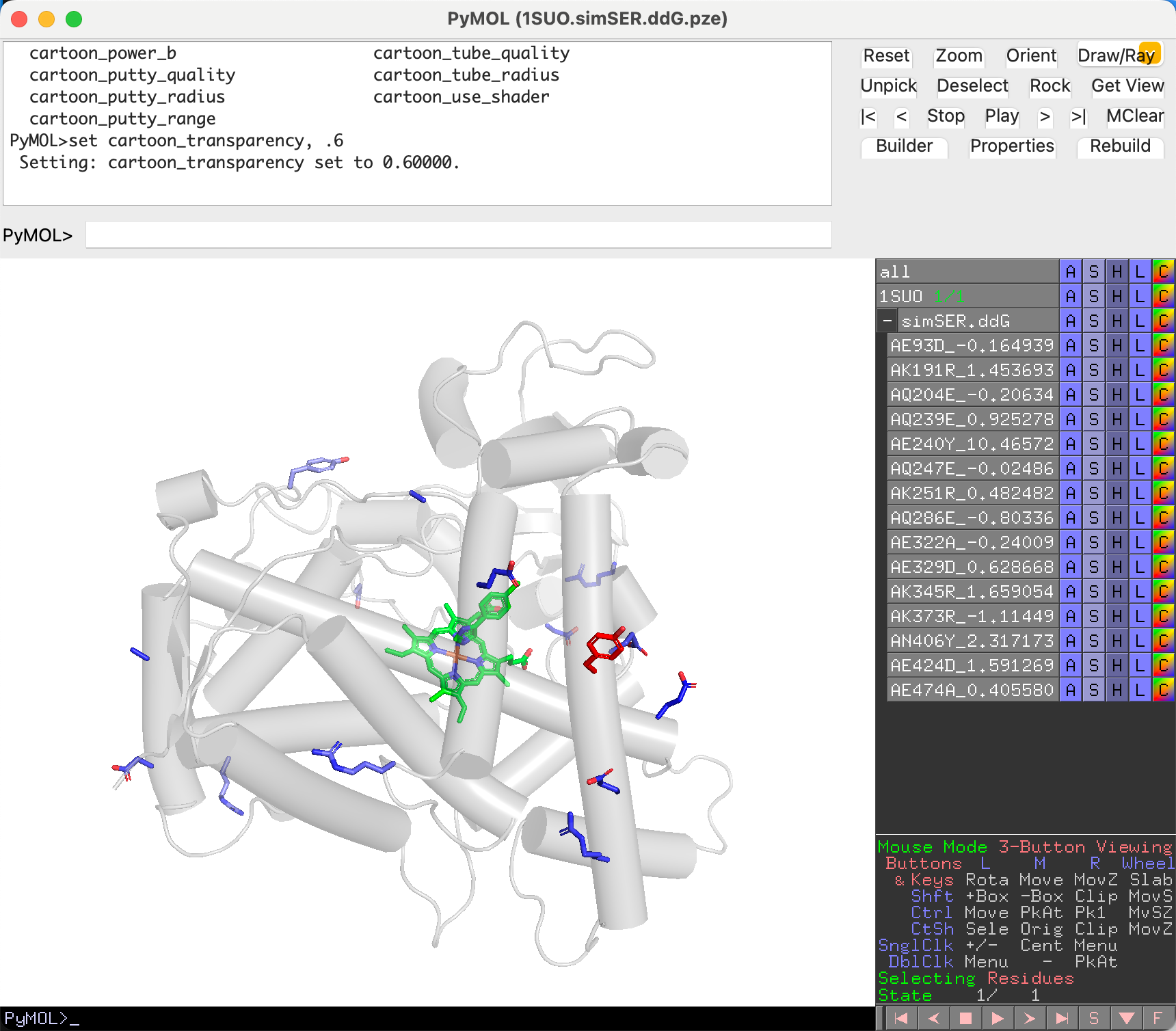
- Click a mutant in the PyMOL viewer to select it.
- Click Hide on the right panel to mark it for removal.
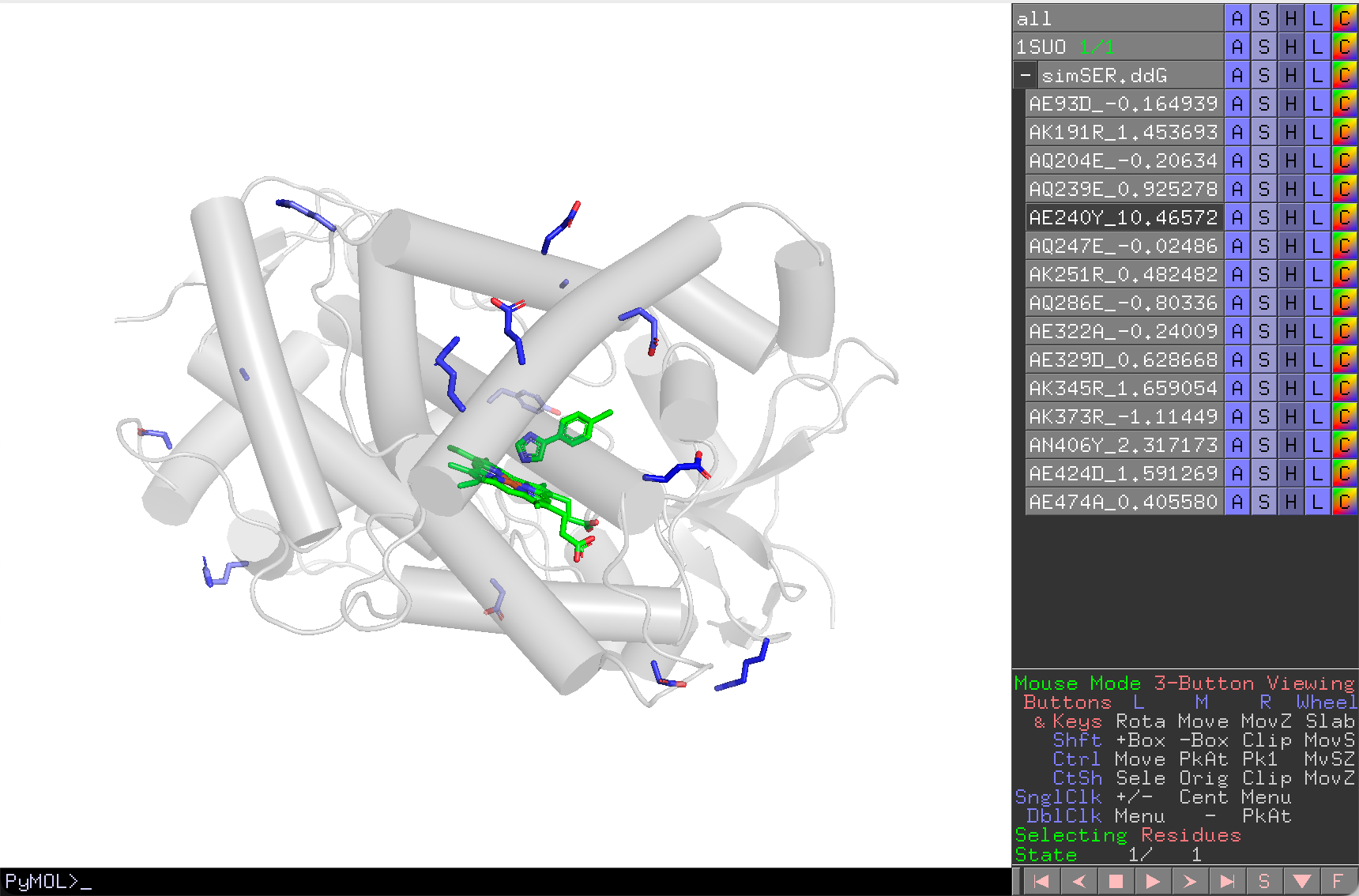
- Click Reduce Session to delete hidden mutants.
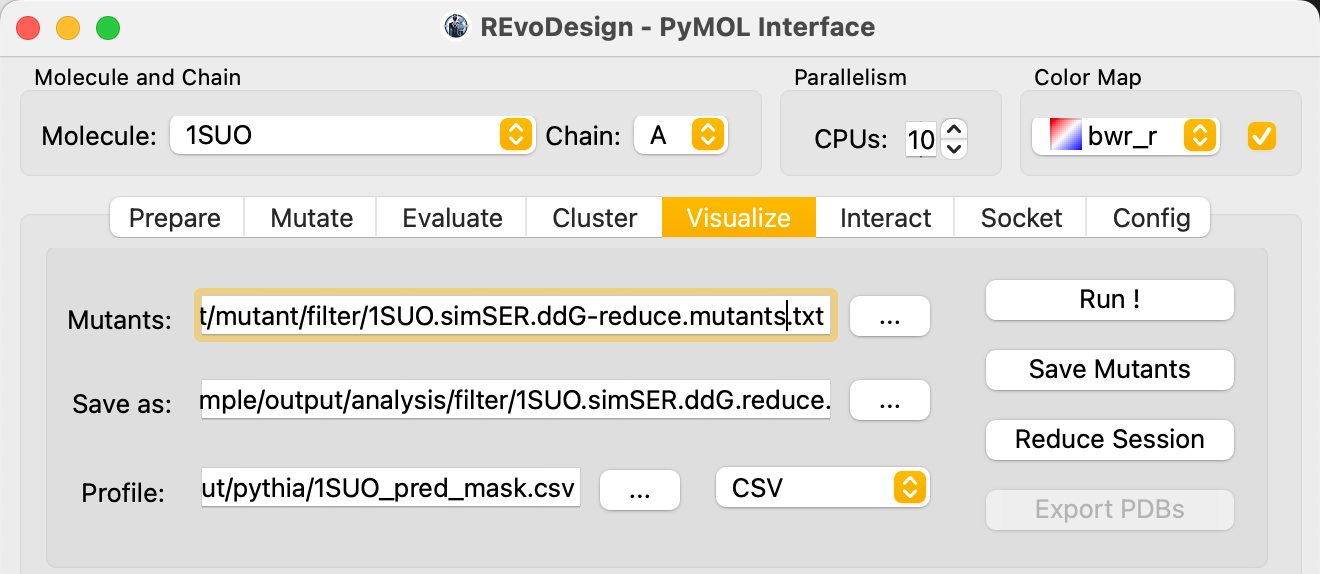
- Rename the mutant table and click Save Mutant to persist.
Displaying Experimental Data on Structure¶
Map your experimental assay results (e.g., enzyme activity, product titer) onto the 3D structure for visual analysis:
-
Prepare a CSV or Excel table:
mutant normalized group WT_1 0 control wt_2 -0.1 control WT 0 control AE93D 0.1 low AK191R 0.2 medium AQ204E 0.3 high -
In the Visualize tab:
- Set Mutants to the CSV path.
- Set Save as to the session save path.
- Clear the Profile path.
- Set Profile type to empty.
- Map column names: Group, Mut, Score to the appropriate CSV column names.
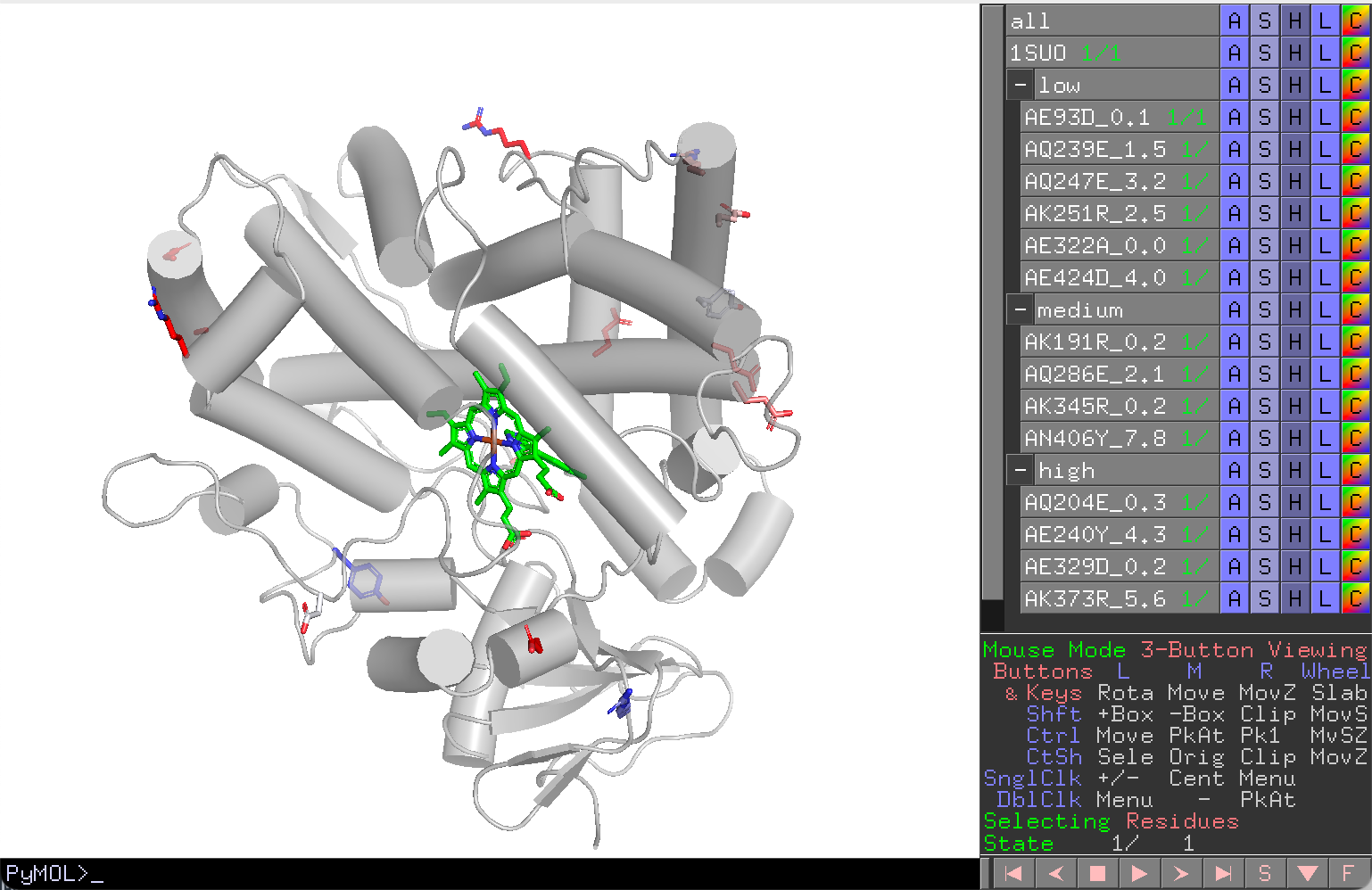
WT handling
Rows whose mutant name contains "WT" (case-insensitive) are treated as controls. Their group assignment is ignored and the WT score is set to the average of all control rows.
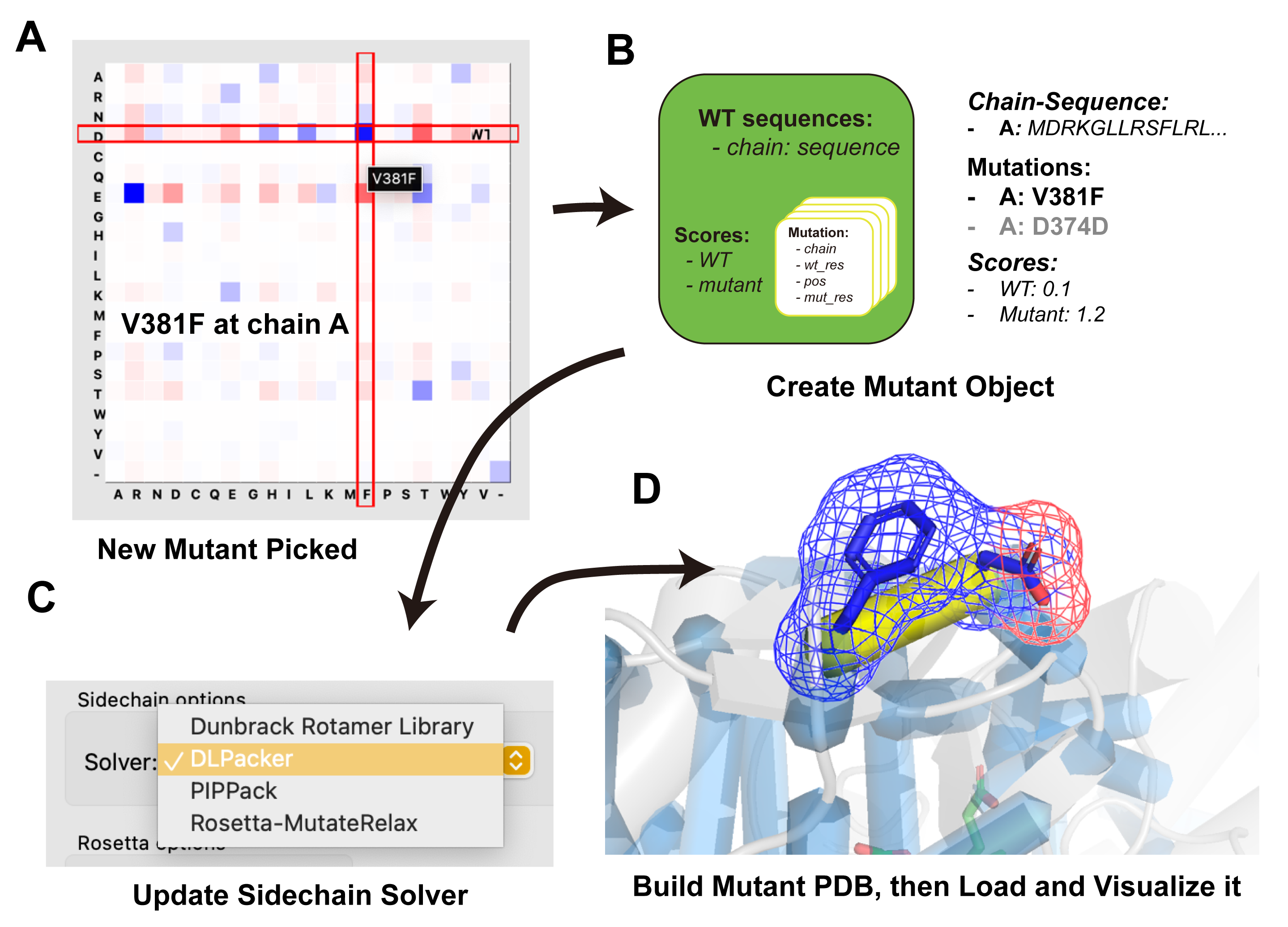
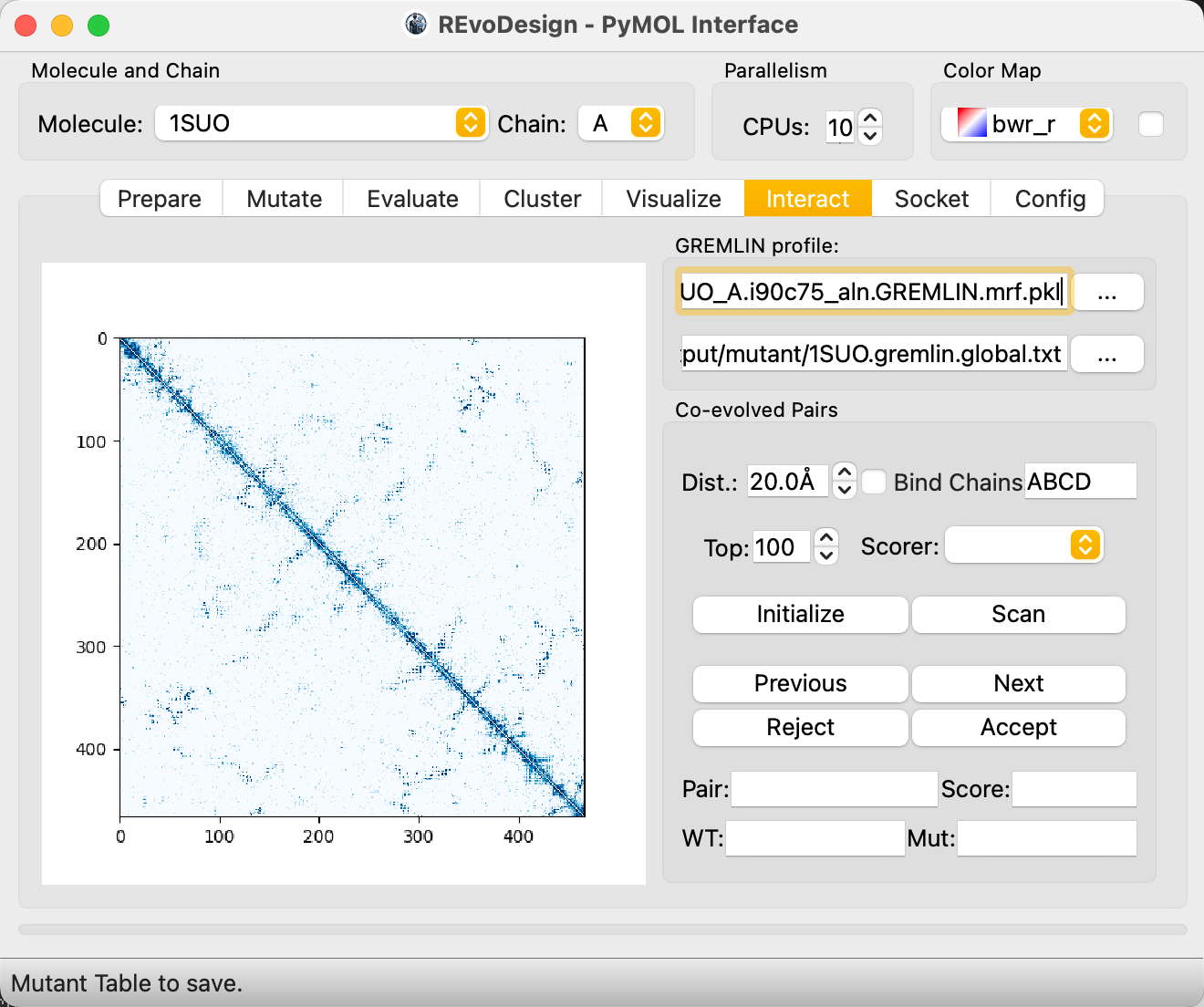
Step 6: Interact — Co-Evolution Analysis¶
The Interact tab uses GREMLIN Markov Random Field (MRF) models to identify co-evolved residue pairs, revealing functional coupling between positions that can guide combinatorial mutation design.
Load GREMLIN Data¶
- In the Interact tab, set the path to the GREMLIN MRF archive
(e.g.,
gremlin_res/1SUO_A.i90c75_aln.GREMLIN.mrf.pkl). - Set the mutant design save path.
- Adjust filters: top N co-evolving pairs, maximum contact distance, homo-oligomer chain binding mode.
- Optionally enable scoring tools for on-the-fly mutant evaluation.
- Click Initialize to load the co-evolution contact map.
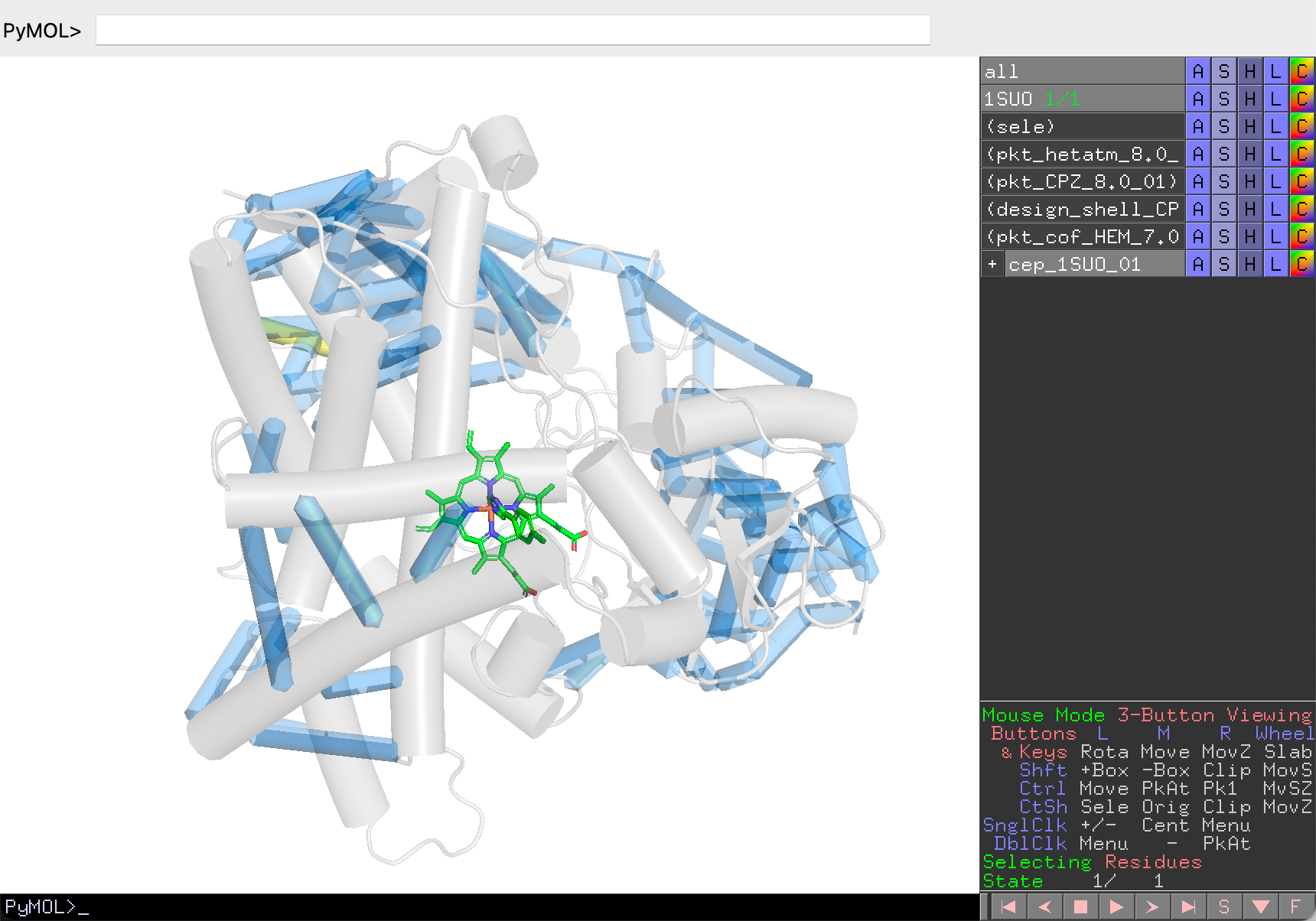
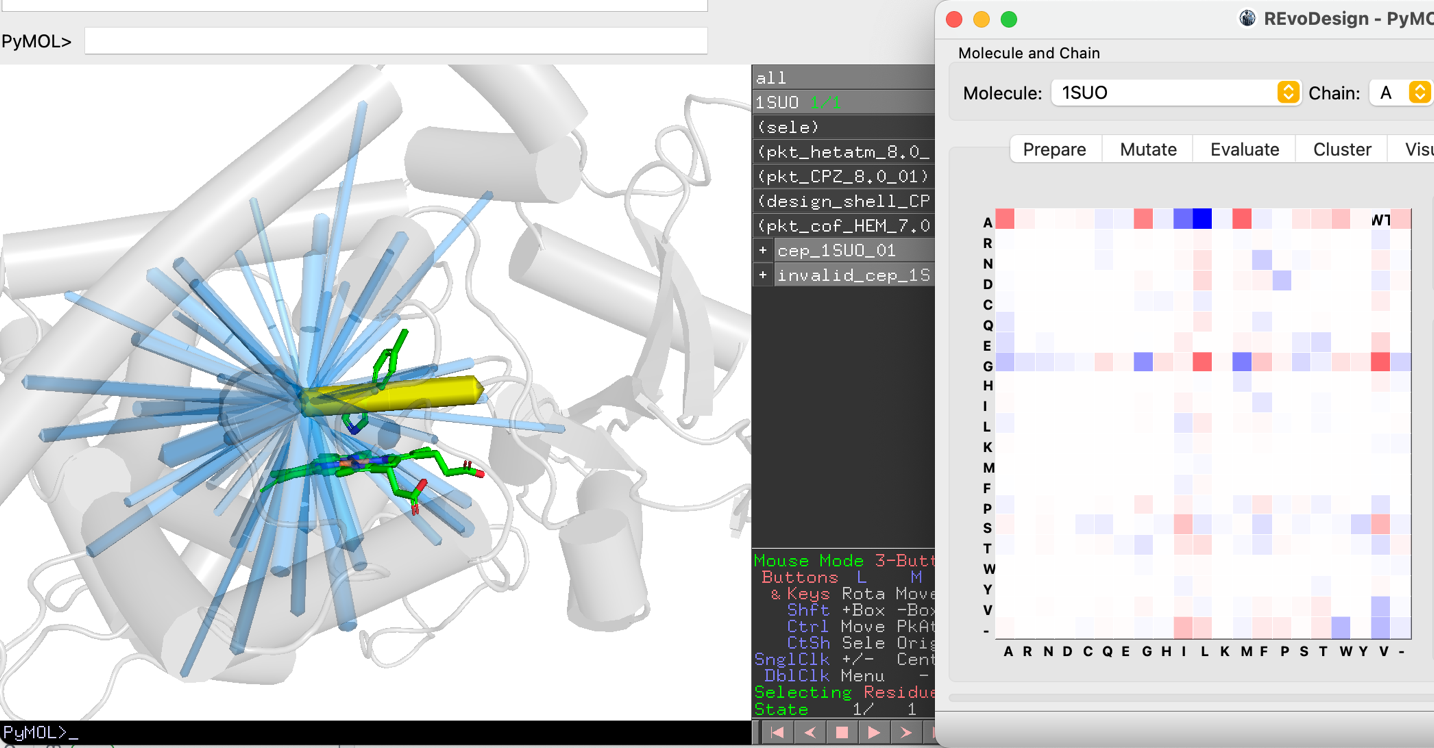
Global Co-Evolution Scan¶
- Click Scan to analyze the top co-evolving pairs within the distance cutoff.
- Results are displayed as backbone traces: blue cylinders represent pairs, a yellow cylinder highlights the current pair. Cylinder thickness indicates co-evolution signal strength.
- Navigate pairs with Previous / Next.
- For each pair, the MRF matrix shows the 20×20 amino acid combination space. Grid cell color represents the GREMLIN probability for that residue pair.
- Click any cell to instantly generate the corresponding double mutant. The mutant flows through: build → sidechain modeling → scoring (if enabled) → grouping → display.
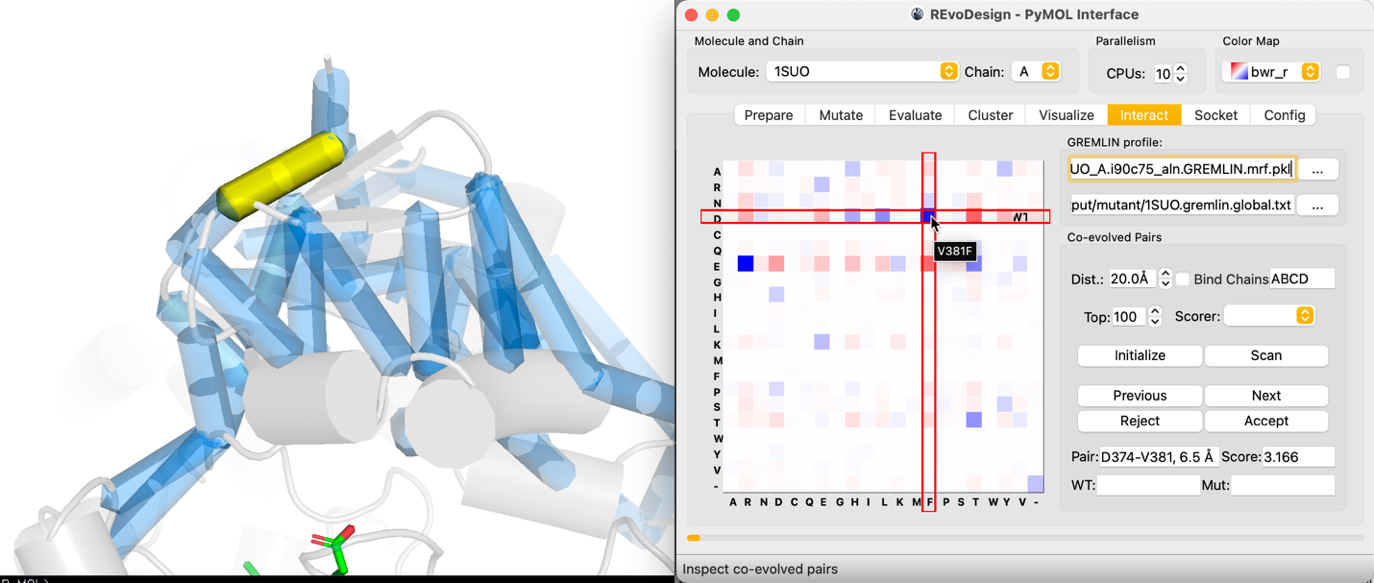
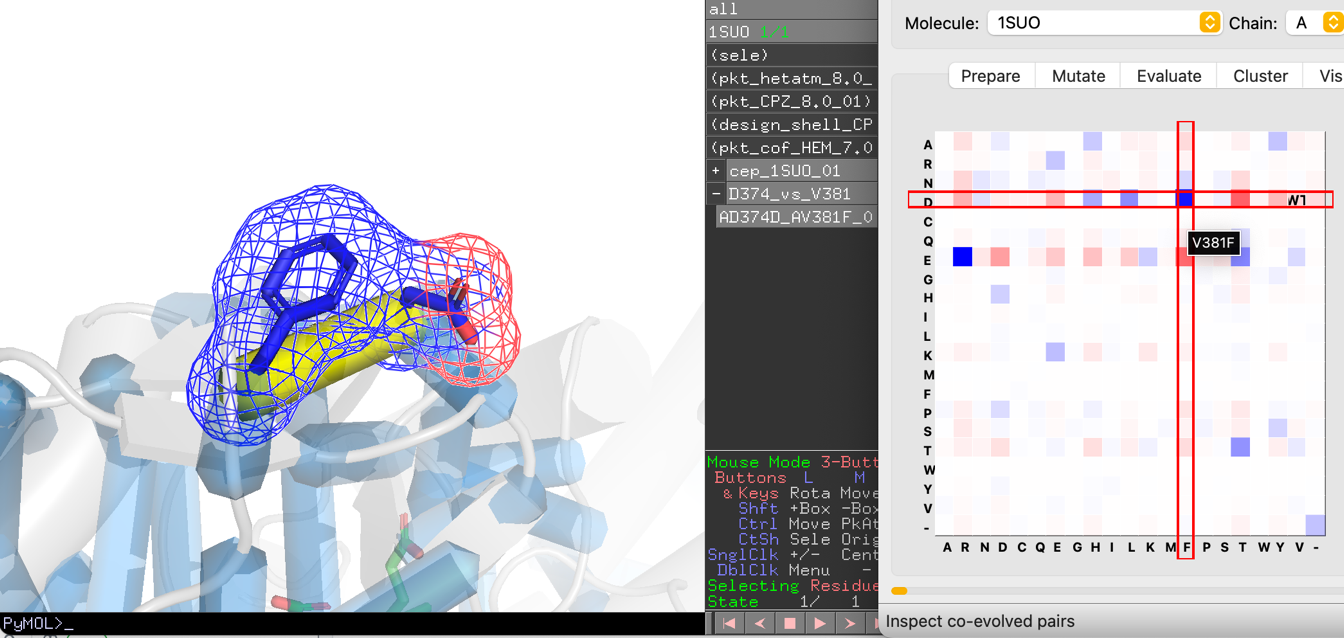
The WT cell marks the wild-type residue combination at the current pair.
Local Co-Evolution Analysis¶
Local analysis focuses on co-evolution partners of a single residue of interest:
- In PyMOL, click on a residue to create a
seleselection object. (Or use:select sele, 1SUO and resi 298) - Ensure the
seleselection is enabled (shown/active in PyMOL). - Click Scan in the Interact tab.
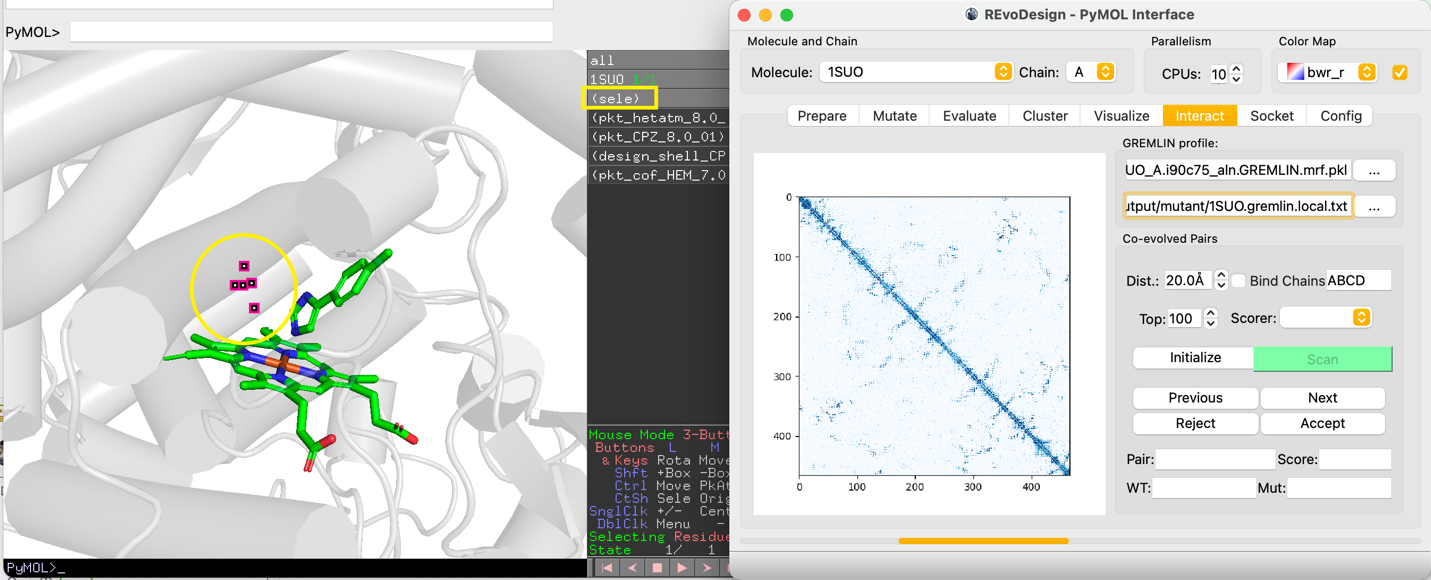
Mutants designed from GREMLIN analysis must be explicitly saved — either click Accept in the Interact tab or switch to the Evaluate tab for structured rational screening.